
27: Síndrome de Prune Belly
Este capítulo levará aproximadamente 28 minutos para ler.
Introdução
A síndrome de Prune Belly (PBS), também denominada síndrome de Eagle-Barrett, é uma condição multissistêmica rara tipicamente caracterizada por uma constelação de anomalias com graus variáveis de gravidade, sendo os principais achados a deficiência da musculatura abdominal, testículos intra-abdominais bilaterais e trato urinário anômalo.1,2Anomalias adicionais associadas não geniturinárias envolvem o trato respiratório, o trato gastrointestinal, o sistema cardíaco e o sistema musculoesquelético.3 A gravidade da doença existe ao longo de um amplo continuum, com algumas crianças não sobrevivendo ao período neonatal e outras sendo minimamente afetadas.4
Embriologia
Diversas teorias predominantes foram propostas sobre a embriogênese da PBS, embora nenhuma delas tenha aceitação universal, e há alguma sobreposição entre elas. As quatro principais teorias são 1, uma obstrução uretral posterior precoce in útero (provavelmente uma próstata hipoplásica/displásica ou uretra anormal), resultando em dilatação vesical proximal, ureteral e renal, associada a desenvolvimento deficiente da parede abdominal 2, um defeito primário no mesoderma da placa lateral, que é o precursor dos uréteres, bexiga, próstata, uretra e gubernáculo 3, um defeito intrínseco do trato urinário levando a dilatação ureteral e ascite fetal e 4, um defeito do saco vitelino.5,6
Epidemiologia
A incidência de PBS foi relatada como sendo de 1 em 29.000 a 1 em 40.000 nascidos vivos, semelhante à da extrofia vesical, com 95% dos pacientes afetados sendo do sexo masculino.1,2,7 As pacientes do sexo feminino representam menos de 5% de todos os casos de PBS e apresentam deficiência da parede abdominal e um trato urinário dilatado e dismórfico, sem qualquer anomalia gonadal associada.8 Observa-se maior incidência em gêmeos, indivíduos negros e crianças nascidas de mães mais jovens.9 Embora a PBS frequentemente se apresente como um evento esporádico, suspeita-se também de contribuição genética, com mutações em genes que codificam fatores de transcrição do músculo liso, filamentos contráteis e enzimas/receptores neurais.10,11
Características Clínicas e Patogênese
Anomalias geniturinárias
Rins
O espectro de anomalias renais em pacientes com PBS se estende de parênquima renal normal a displasia. Cerca de metade dos casos apresenta displasia renal, geralmente das variedades tipo II e IV de Potter, e isso pode variar entre os dois lados também.1,2 Ao passo que a variedade tipo II, com poucos néfrons e desorganização parenquimatosa, é mais indicativa de um defeito mesenquimal renal, a variedade tipo IV, com cistos corticais e tubulares, está associada à obstrução de saída, em que não houve descompressão do úraco.12 Embora o sistema coletor renal seja caracteristicamente dilatado, muitas vezes em grau acentuado, o grau de dilatação, porém, não se correlaciona com o grau de displasia renal, e a morfologia calicial pode estar bem preservada, mesmo na presença de dilatação maciça. Embora a hidronefrose geralmente seja não obstrutiva, pode ocorrer também obstrução da junção ureteropélvica primária ou secundária. Entretanto, é importante notar que é a infecção renal, e não a obstrução, que representa o maior risco para a função renal nesses pacientes.1,2
Ureteres
Os ureteres são tipicamente dilatados, tortuosos e redundantes. Embora a dilatação maciça e a estenose possam ocorrer em todos os níveis, as porções proximais (superiores) dos ureteres são geralmente menos anormais do que os segmentos distais. O refluxo vesicoureteral (RVU) está presente em 75% das crianças com PBS.12 A obstrução não é comum, mas foi relatada nas junções ureteropélvica e ureterovesical. Cortes histológicos demonstram escassez de células musculares lisas e aumento da relação colágeno/músculo liso nos segmentos distais mais afetados, especialmente nos refluxantes, com células musculares lisas de aspecto mais normal nos segmentos proximais.13 Esse fato é crítico quando se realiza reconstrução ureteral. Acredita-se que a diminuição do número de miofibrilas grossas e finas observada no exame ultraestrutural contribua para a peristalse ineficaz devido à coaptação deficiente da parede ureteral, resultando em estase do trato urinário superior, o que pode levar à infecção. Além disso, em muitos pacientes, a gravidade das anomalias do trato urinário não é proporcional ao grau de flacidez da parede abdominal.14
Bexiga
A bexiga geralmente se apresenta maciçamente aumentada, com um pseudodivertículo no úraco e úraco pérvio em 25% a 30% dos pacientes.12 Diferentemente do quadro observado em bexigas obstruídas por outras causas , apesar de muito espessa, o contorno vesical é liso. Durante a micção, o colo vesical se abre amplamente em uma uretra prostática dilatada.1,2 Histologicamente, na ausência de obstrução, a bexiga apresenta uma relação aumentada entre colágeno e fibras musculares, contudo pode-se observar hipertrofia do músculo liso quando há obstrução. Na cistoscopia , o trígono está alargado, com os orifícios ureterais deslocados superior e lateralmente , o que possivelmente contribui para a alta incidência de refluxo vesicoureteral. Achados característicos em uma avaliação urodinâmica incluem uma bexiga com complacência normal, com primeira sensação de micção tardia e grande capacidade.15 A eficiência miccional dessas bexigas é variável, com algumas esvaziando completamente, enquanto outras apresentam resíduo pós-miccional significativo, possivelmente devido a uma obstrução relativa da saída e à incapacidade do músculo detrusor de gerar pressão suficiente durante a micção nesses pacientes.16 O termo micção “unbalanced” é geralmente usado para se referir à situação anterior em que uma resistência relativa ao fluxo de saída impede o esvaziamento eficaz da bexiga.1,2 Embora 50% dos pacientes com síndrome de prune-belly urinem espontaneamente com pressões miccionais normais, taxas de fluxo normais e baixos resíduos pós-miccionais, observou-se que a deterioração da micção balanceada pode ocorrer com o crescimento, resultando em resíduos pós-miccionais significativos, enfatizando a necessidade de avaliação periódica.16
Próstata e Órgãos Sexuais Acessórios
A dilatação uretral posterior deve-se à hipoplasia prostática, provavelmente relacionada a um desenvolvimento mesenquimal-epitelial anormal.12,13,14,15,16,17,18,19 Cortes histológicos revelam poucos elementos celulares prostáticos, com redução de células epiteliais e de músculo liso e aumento de células do tecido conjuntivo.19 Observou-se que cerca de 20% dos pacientes também apresentam lesões obstrutivas na uretra posterior distal, na forma de atresia uretral, válvulas, estenose uretral, membrana uretral e divertículo uretral. A falta de tecido parenquimatoso prostático pode levar a uma angulação da uretra durante a micção, a qual foi denominada por Stephens de válvula tipo IV.12 A hipoplasia prostática e o colo vesical incompetente associado também são considerados fatores na falha ejaculatória/ ejaculação retrógrada em pacientes com PBS.1,2 Os órgãos sexuais acessórios, como os ductos deferentes e as vesículas seminais, são frequentemente atrésicos, embora qualquer um deles possa estar dilatado ou espessado. O epidídimo pode estar mal aderido ao testículo e pode haver também falta de continuidade entre os dútulos eferentes e a rede testicular.20
Uretra Anterior
Embora a uretra anterior da criança com PBS geralmente seja normal, várias anomalias do segmento uretral foram relatadas; as mais comuns são atresia ou hipoplasia uretral e megalouretra.21,22,23,24,25 Postula-se que a atresia ou hipoplasia uretral ocorra porque a uretra não é utilizada, em vez de malformada. A atresia uretral é frequentemente letal se não estiver associada à descompressão na forma de úraco pérvio ou, às vezes, até à ruptura espontânea da bexiga com formação de fístula.22
Uma característica singular na PBS é a ocorrência de megalouretra.26 Tem sido proposta como causa de megalouretra uma obstrução transitória in útero da junção entre a uretra glandular e a peniana. Duas variações de megalouretra são observadas em pacientes com PBS — a saber, a fusiforme (Figura 1) e a escafoide (Figura 2), (Figura 3) variante.24 O tipo fusiforme corresponde a uma deficiência dos corpos cavernosos, assim como do corpo esponjoso, provavelmente resultante de uma deficiência mesenquimal das pregas uretrais, e é observado clinicamente como dilatação de todo o falo durante a micção. Por outro lado, a variante escafoide, que provavelmente decorre de uma deficiência mesenquimal dos tecidos de suporte da uretra, levando a deficiência apenas do corpo esponjoso com preservação da glande e dos corpos cavernosos, caracteriza-se por dilatação da uretra ventral durante a micção.26
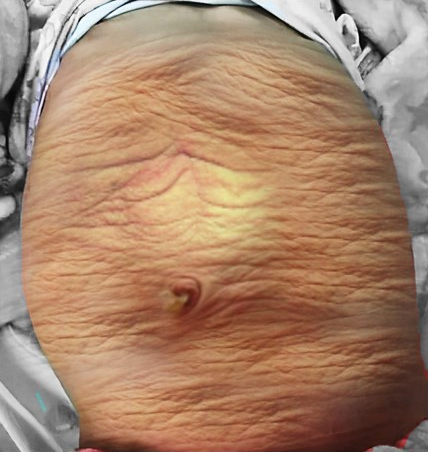
Figura 1 Fotografia clínica de uma criança com síndrome do abdome em ameixa seca
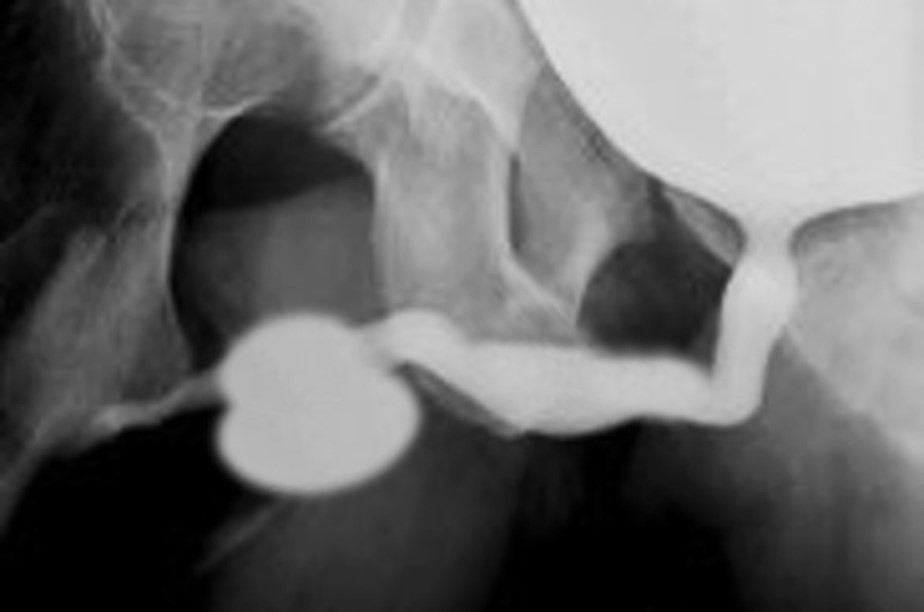
Figura 2 Megalouretra fusiforme
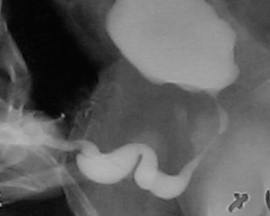
Figura 3 Megalouretra escafoide
Testículos
Testículos intra-abdominais bilaterais situados sobre os vasos ilíacos e adjacentes aos ureteres dilatados são considerados um dos achados típicos na PBS.1,2 Embora forças mecânicas, como obstrução mecânica por megacistose e baixa pressão intra-abdominal devido a anomalias da parede abdominal, tenham sido há muito tempo postuladas como a etiologia para essa falha da descida testicular, o fato de que alguns pacientes com as anomalias típicas do trato urinário e da musculatura abdominal podem ter testículos descendidos levanta alguma dúvida acerca de fatores puramente mecânicos.1,2 No que diz respeito a se os testículos em crianças com PBS são intrinsecamente diferentes daqueles sem PBS, Pak et al compararam a histologia de testículos intra-abdominais e não encontraram diferença nas contagens de células germinativas, espermatogônias e células de Leydig entre testículos com PBS e testículos fetais intra-abdominais sem PBS.27,28 No entanto, Orvis et al observaram redução do número de espermatogônias e hiperplasia de células de Leydig em testículos fetais com PBS, sugerindo uma anomalia testicular intrínseca.29 Quanto ao risco de malignidade nesses testículos, embora o risco possa ser relativamente baixo devido à falta de epitélio germinativo, a orquidopexia precoce e o seguimento em longo prazo são necessários para reduzir o risco de malignidade testicular e aprimorar a detecção.30,31
Defeito da Parede Abdominal
A aparência da parede abdominal é uma característica marcante em crianças com PBS. Mais comumente, a deficiência muscular é irregular e em mosaico, com os segmentos musculares medial e inferior tipicamente os mais deficientes e podendo ser desproporcional às anomalias do trato urinário. As áreas mais gravemente afetadas podem ter pele, tecido adiposo subcutâneo e uma única camada fibrosa sobre o peritônio, com exame histológico por microscopia eletrônica demonstrando um padrão inespecífico de desarranjo de miofilamentos, desorganização das linhas Z e proliferação mitocondrial.32,33 Apesar dessas alterações da parede abdominal, observa-se nessas crianças boa cicatrização, sem tendência a infecções ou hérnias incisionais. O aspecto típico ao nascimento é o de pele enrugada e redundante, com um abdome com abaulamento nos flancos. À medida que os pacientes crescem, alguns mostram melhora do tônus e do aspecto abdominal, com menos rugas, ao passo que em outros o abdome adquire mais o aspecto de abdome globoso.1,2 A marcha geralmente não é afetada, embora possa ser atrasada, e as crianças tendem a rolar para o lado e usar os braços para sentar a partir do decúbito dorsal. O fraco suporte da parede torácica inferior pode levar ao alargamento dos arcos costais e a consequente redução da eficácia da tosse torna essas crianças mais vulneráveis a doenças respiratórias.
Anomalias extrageniturinárias
De todas as crianças com PBS, 75% apresentam anomalias extraurinárias.1,2,4 Após o evidente defeito da parede abdominal, os problemas mais comuns são cardíacos, pulmonares e ortopédicos (Tabela 1).34,35,36,37,38,39,40 Além dessas morbidades específicas de órgão, quase 50% das crianças com PBS nascem prematuras, o que contribui significativamente para comorbidades.
Tabela 1 Anomalias extrageniturinárias comuns observadas na síndrome do abdome em ameixa seca
| Sistema | Defeitos comuns | Observações |
|---|---|---|
| Anomalias Cardíacas (10%) | Persistência do canal arterial, defeitos do septo atrial e ventricular, defeito do septo ventricular e tetralogia de Fallot | As anomalias cardíacas ao nascimento podem ter precedência sobre as questões urológicas. |
| Anomalias Pulmonares (55%) | Hipoplasia pulmonar Pneumotórax e pneumomediastino podem ser observados com ou sem hipoplasia pulmonar | A hipoplasia pulmonar pode resultar de oligohidrâmnio grave relacionado à displasia renal ou obstrução grave da via de saída vesical e pode resultar em óbito do recém-nascido. Em quase metade dos recém-nascidos com PBS, serão necessários intubação e suporte ventilatório mecânico, com suas morbidades associadas. A incapacidade de gerar pressão intra-abdominal significativa e as anomalias musculoesqueléticas associadas, como escoliose e alterações da caixa torácica, mais comumente pectus excavatum, podem contribuir para aumento do risco de pneumonia e atelectasia lobar nessas crianças . |
| Anomalias Gastrointestinais (30%) | As anomalias resultam de rotação incompleta do intestino médio, dando origem a um mesentério amplo, o que resulta em maior mobilidade intestinal com malrotação intestinal, vólvulo, atresias e estenoses. Torção esplênica relacionada à fixação mesentérica anormal também foi relatada. Onfalocele, gastrosquise, situs inversus abdominis, VACTERL (defeitos vertebrais, atresia anal, defeitos cardíacos, fístula traqueoesofágica, anomalias renais e anomalias dos membros) e anomalias anorretais foram relatadas. | Com a capacidade limitada de gerar pressão intra-abdominal em decorrência da hipoplasia da musculatura abdominal, a constipação torna-se um problema para toda a vida e pode levar a megacólon adquirido. |
| Anomalias Ortopédicas (65%) | Fossetas da face lateral dos joelhos são um achado comum no oligohidrâmnio e são frequentemente vistas em pacientes com PBS, juntamente com outros defeitos como pé torto equinovaro (26%), displasia do quadril (5%) e escoliose congênita (4%). Hipoplasia, ausência ou amputação dos membros inferiores. | Tendo em vista que a maioria desses defeitos musculoesqueléticos é unilateral, a causa mais provável desses defeitos é o efeito compressivo do oligohidrâmnio. Outro mecanismo proposto para as anomalias dos membros inferiores tem sido a compressão dos vasos ilíacos externos pela bexiga distendida, comprometendo assim o suprimento sanguíneo para os membros inferiores Também, algumas dessas malformações, como osteodistrofia renal, luxação do quadril, escoliose e pectus excavatum ou carinatum, tendem a se manifestar ou piorar com o crescimento. |
Diagnóstico e Avaliação
Pré-natal
Embora o diagnóstico de PBS por ultrassonografia tenha sido relatado já às 11-12 semanas de gestação, a acurácia diagnóstica varia de 30% a 85%, pois a PBS se apresenta no pré-natal com muitos achados ao ultrassom comparáveis aos da obstrução do trato urinário inferior (LUTO) por outras causas, incluindo válvulas uretrais posteriores, ureterocele e atresia uretral.40,41,42,43,44 O diagnóstico pré-natal de PBS deve ser considerado sempre que as seguintes anomalias ao ultrassom forem claramente identificadas: oligohidrâmnio, anormalidades urinárias (dilatação do trato urinário, megacistia, hidroureteronefrose bilateral) e ausência de musculatura abdominal. O diagnóstico precoce não apenas melhora a sobrevida ao levar o médico assistente a realizar o parto em um centro terciário para manejo multidisciplinar imediato dos recém-nascidos afetados, como também oferece aos pais expectantes a opção de interrupção voluntária da gestação, se desejado.43
Recém-nascido
A aparência da parede abdominal é diagnóstica no recém-nascido, independentemente de o diagnóstico ter sido conhecido no período pré-natal. Crianças afetadas por PBS representam um desafio único e exigem cuidados multidisciplinares imediatos por uma equipe composta por neonatologista, urologista, nefrologista, bem como cardiologista e ortopedista pediátrico quando indicado. Dada a ampla variação da gravidade da doença, uma avaliação multissistêmica para excluir malformações cardíacas, pulmonares ou outras associadas deve ocorrer no período neonatal. É necessária uma radiografia de tórax imediata para excluir pneumotórax e pneumomediastino associados. Como ocorre com LUTO, a evolução pós-natal inicial é ditada pela gravidade das comorbidades, como hipoplasia pulmonar, com piores desfechos nos prematuros e naqueles com hipoplasia pulmonar grave.45,46,47
O oligoidrâmnio antenatal nesses recém-nascidos indica função renal comprometida. As dosagens iniciais de creatinina refletem a função renal materna e coletas repetidas são necessárias. Se após 48–72 horas a creatinina sérica for maior que 1.0 no recém-nascido a termo ou maior que 1.5 no prematuro, há um grau de insuficiência renal. Um aumento progressivo da creatinina sérica nas primeiras semanas de vida, prenuncia um desfecho desfavorável. Se o nadir inicial da creatinina for menor que 0.7 mg/dL, então a insuficiência renal subsequente é improvável.48 A urina é enviada para cultura inicial e inicia-se profilaxia antibiótica. A dosagem de eletrólitos séricos e urinários ao nascimento pode ser útil na avaliação da conservação de sódio, implicando função renal adequada.
Avaliação Diagnóstica
Uma avaliação urológica completa é realizada após a estabilização do paciente devido a outras comorbidades. Exame físico e ultrassonografia do trato urinário são inicialmente necessários.46 As ultrassonografias renais fornecem informações sobre a espessura cortical, a presença de alterações císticas e o tamanho renal. Exames ultrassonográficos antes e após a micção fornecem indícios da presença de refluxo vesicoureteral e de resíduo urinário pós-miccional.
Inicialmente, realiza-se uma cistouretrografia miccional (VCUG) naqueles lactentes com função renal anormal para assegurar que não esteja presente uma obstrução uretral anatômica verdadeira. Esta “variante letal” e a potencial confusão com válvulas uretrais posteriores “simulando” a síndrome do abdome em ameixa seca devem ser diagnosticadas, pois o tratamento muda dramaticamente. Além disso, a VCUG demonstra refluxo vesicoureteral em até 85% dos pacientes e avalia o esvaziamento vesical. Todos os lactentes são tratados com antibióticos antes da cateterização estéril para reduzir o risco de infecção. Em geral, a avaliação radiológica do trato urinário é ajustada conforme a filosofia de manejo do cirurgião urológico individual. Estudos contrastados que exigem instrumentação arriscada são evitados naqueles lactentes com boa função renal.22
Após a avaliação inicial, uma cintilografia renal com mercaptoacetiltriglicina (MAG3) pode fornecer informações tanto funcionais quanto anatômicas. O fluxo sanguíneo, a função renal diferencial e a drenagem em resposta à furosemida (Lasix) podem ser avaliados e comparados com estudos subsequentes. As limitações da renografia diurética em demonstrar obstrução em pacientes com síndrome de prune-belly foram observadas.49 O teste de Whitaker é ocasionalmente necessário como o estudo definitivo para demonstrar obstrução significativa, embora isso seja em grande parte de importância histórica. A cintilografia renal com ácido dimercaptossuccínico (DMSA) é realizada em vez de urograma excretor (IVP) para avaliar o tamanho renal, a massa tubular e a função diferencial. Estudos comparativos são inestimáveis para avaliar o crescimento renal e as cicatrizes, pois esses pacientes requerem acompanhamento a longo prazo. Alguns grupos têm defendido a urografia por ressonância magnética (MRU) nesses pacientes devido à sua alta resolução e a excelentes dados funcionais e anatômicos que permitem visualização detalhada das anomalias do trato superior, o que também pode ser útil no planejamento operatório.50 Como mostrado na Tabela 1, há três categorias principais de apresentação neonatal conforme descrito por Woodard.1,2
Manejo e Desfechos
Manejo Pré-natal
Intervenção intrauterina
Com a melhora das técnicas avançadas de imagem e diagnóstico materno-fetais, a síndrome de Prune Belly pode ser suspeitada por imagem precocemente na gestação. A intervenção fetal geralmente visa tratar o oligoidrâmnio ou anidrâmnio, que está associado à hipoplasia pulmonar, a principal causa de mortalidade perinatal. Os critérios para intervenção intrauterina incluem gestação no segundo ou terceiro trimestre, oligoidrâmnio grave, megacistia, hidronefrose avançada, cariótipo normal, extensão limitada de outras anomalias congênitas detectadas e análises seriadas favoráveis da urina fetal aspirada.51,52,53 Embora existam vários relatos de procedimentos de derivação vesicoamniótica para uropatia obstrutiva fetal, as revisões não conseguiram documentar um efeito benéfico da intervenção fetal sobre a função renal subsequente e tampouco a função pulmonar pode ser assegurada, apesar do restabelecimento de níveis normais de líquido amniótico. Como a maioria dos pacientes com síndrome de Prune Belly não apresenta evidência demonstrável de obstrução e a função renal não se correlaciona com o grau de dilatação presente no trato urinário, os riscos e benefícios da intervenção intrauterina devem ser ponderados cuidadosamente.53
Manejo pós-natal
Os objetivos gerais do manejo são preservar a função renal e prevenir infecção. O alcance desses objetivos é possível por meio de uma variedade de abordagens terapêuticas, que vão desde conduta expectante até reconstrução cirúrgica imediata do trato urinário. Evita-se a intervenção cirúrgica precoce no recém-nascido, a menos que ocorra elevação da creatinina ou infecção, o que requer vesicostomia precoce. Como mostrado na Tabela 2, o manejo nesses pacientes depende da categoria de gravidade na apresentação.
Tabela 2 Apresentação e manejo de diferentes categorias com base nos critérios de Woodward de apresentação neonatal
| Categoria de gravidade | Apresentação | Manejo | Acompanhamento e desfechos |
|---|---|---|---|
| I | Oligohidrâmnio acentuado secundário a displasia renal e/ou obstrução da via de saída, resultando em hipoplasia pulmonar grave e anomalias esqueléticas | Naqueles poucos lactentes que sobrevivem à agressão pulmonar, tem-se tentado derivação urinária alta por pielostomia cutânea; no entanto, a recuperação da função renal geralmente não é possível em razão da grave displasia renal subjacente. | Esses neonatos frequentemente vão a óbito em poucos dias e as intervenções são limitadas. Associados a oligohidrâmnio pré-natal e hipoplasia pulmonar pós-natal, geralmente morrem no período pós-natal imediato devido a complicações pulmonares. |
| A fácies de Potter pode estar presente e é secundária ao oligohidrâmnio. As características incluem micrognatia, olhos muito separados, fendas palpebrais achatadas, epicanto proeminente, ponte nasal achatada, orelhas de implantação baixa desprovidas de cartilagem e deformidades esqueléticas. | Pode ser necessária apenas cateterização | ||
| Casos de atresia uretral são tipicamente desta categoria mais grave. | |||
| II | Insuficiência renal moderada e hidroureteronefrose moderada a grave. Hipoplasia pulmonar não é uma característica proeminente. | Essas crianças recebem antibióticos profiláticos de longo prazo e são monitoradas seriadamente quanto a infecções e qualquer redução da função renal. | Necessária vigilância pós-operatória por toda a vida para avaliar a função renal e a drenagem ideal dos tratos superior e inferior com estudos funcionais e anatômicos. |
| Realiza-se reconstrução agressiva do trato urinário na presença de infecção documentada, falha de crescimento renal adequado ou declínio da função renal. | |||
| Entretanto, a reconstrução cirúrgica geralmente é adiada até que o lactente tenha 3 meses de idade ou mais para permitir a maturação pulmonar. | |||
| Se a insuficiência renal for grave ou houver infecção inicial, recomenda-se um período de derivação urinária por meio de vesicostomia cutânea. | |||
| Ureterostomias cutâneas provavelmente acrescentam pouco para melhorar a drenagem. Além disso, o ureter superior é anatomicamente e histologicamente o mais normal e deve ser preservado caso se opte posteriormente por uma reconstrução ajustada. | |||
| III | Características leves da tríade ou formas incompletas | Constituem a maioria dos pacientes e demonstram boa função renal, apesar da dilatação acentuada do trato urinário superior e inferior. | Semelhante à categoria 2, é necessária vigilância por toda a vida. |
| A função renal é tipicamente normal ou discretamente comprometida e não há insuficiência pulmonar. | A intervenção cirúrgica precoce para reconstruir o trato urinário geralmente não é necessária e esses pacientes são manejados com profilaxia antibiótica de longo prazo e avaliação seriada da função renal por técnicas de imagem seletivas. | O desenvolvimento de infecções urinárias ou a diminuição da função renal impõe avaliação específica adicional sob a forma de avaliação urodinâmica ou renograma diurético para documentar obstrução do trato inferior ou superior que possa necessitar de intervenção. | |
| Esses pacientes se beneficiam de orquidopexia precoce e abdominoplastia, pois a correção da parede abdominal pode melhorar a defecação e a eficiência da micção. |
Intervenções Cirúrgicas na Síndrome de Prune Belly
Trato urinário
Um achado característico na PBS é a dilatação de baixa pressão de todo o trato urinário, que se estende desde a pelve renal proximalmente até a uretra distalmente. A bexiga é tipicamente aumentada e hipotônica, com complacência aumentada e refluxo vesicoureteral (RVU) de baixa pressão presente em aproximadamente 75% dos pacientes. Embora essas bexigas complacentes de grande volume permitam armazenamento em baixa pressão, elas frequentemente apresentam esvaziamento incompleto secundário à contratilidade detrusora reduzida. Portanto, a intervenção urológica inicial é direcionada à drenagem vesical para evitar infecção do trato urinário (ITU) e, assim, preservar a função renal. Dessa forma, circuncisão, antibióticos profiláticos e micção programada/dupla são frequentemente implementados para reduzir a incidência de ITU e proteger as vias urinárias superiores de novas agressões.54,55,56,57 Quando tal manejo conservador falha em prevenir infecção e alcançar esvaziamento adequado, essas crianças podem necessitar de cistoplastia redutora e /ou apendicovesicostomia para facilitar o cateterismo intermitente limpo e/ou cirurgia antirrefluxo para o manejo do RVU associado.58,59,60
Parede Abdominal
A reconstrução da parede abdominal, na qual a porção periférica mais normal da parede abdominal é avançada para sustentar essa porção central anormal e deficiente, é uma consideração importante no manejo cirúrgico de pacientes com PBS. Além da estética, a abdominoplastia também demonstrou melhorar a dinâmica funcional vesical independentemente da reconstrução geniturinária. Testes urodinâmicos são frequentemente realizados no pré-operatório para determinar a necessidade de quaisquer procedimentos vesicais concomitantes, como apendicovesicostomia nos casos de esvaziamento incompleto e/ou reimplante ureteral quando há VUR. Tradicionalmente, três procedimentos cirúrgicos, com suas modificações, foram relatados na literatura para correção dos defeitos da parede abdominal associados à PBS.61,62,63 Enquanto o procedimento de Randolph envolve a excisão de uma porção da parede abdominal inferior para corrigir a redundância fascial vertical, tanto o procedimento de Monfort (Figura 4), (Figura 5), (Figura 6), (Figura 7), (Figura 8) quanto o de Ehrlich utilizam a sobreposição vertical da fáscia para corrigir a redundância lateral, juntamente com o fortalecimento da parede abdominal. Uma abordagem laparoscópica para a abdominoplastia também foi relatada, mostrando-se benéfica tanto ao auxiliar na própria reconstrução quanto ao delinear os variados graus de deficiência muscular.64 Observa-se também que pacientes com PBS podem apresentar características únicas quanto à variação da gravidade e do padrão de deficiência da musculatura abdominal, como uma área específica com ausência completa de músculo. Smith e colegas relataram recentemente suas adaptações ao procedimento de Monfort, que permitem uma correção mais individualizada da laxidade da parede abdominal e o posicionamento do umbigo em uma localização anatomicamente mais correta.
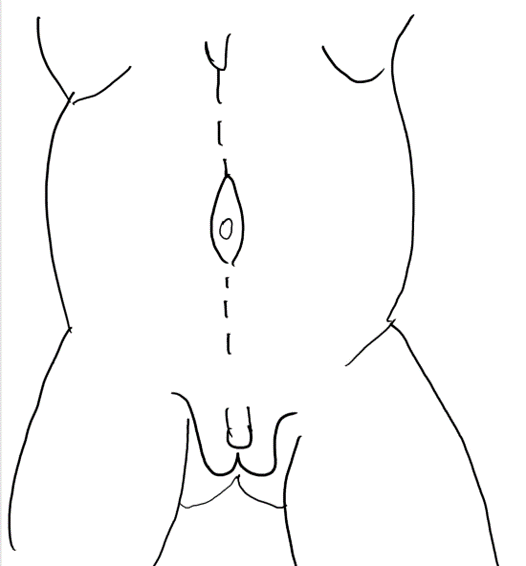
Figura 4 Uma incisão mediana do processo xifoide ao púbis circundando a cicatriz umbilical.
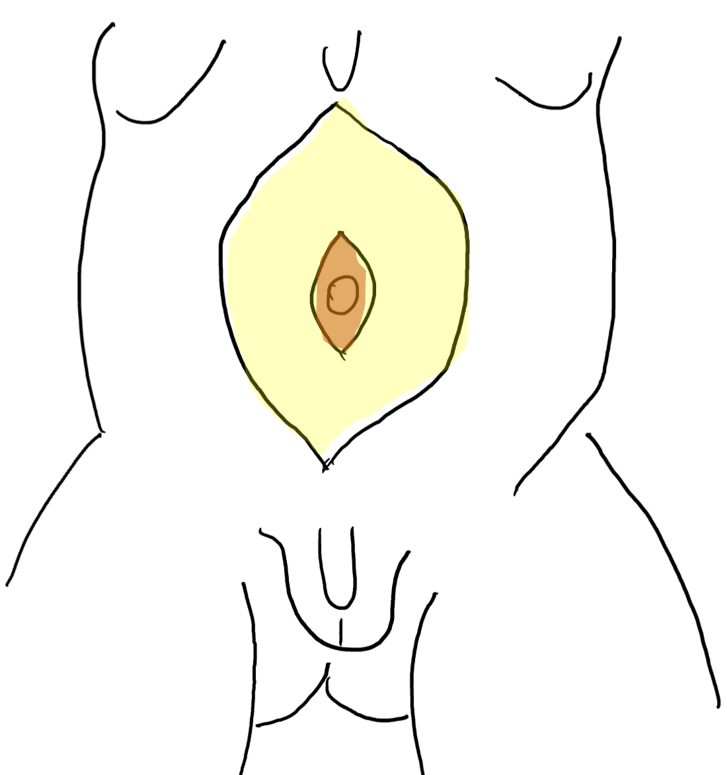
Figura 5 Elevação do retalho cutâneo entre a gordura subcutânea e a camada fascial. O umbigo é sustentado pela faixa fascial central.
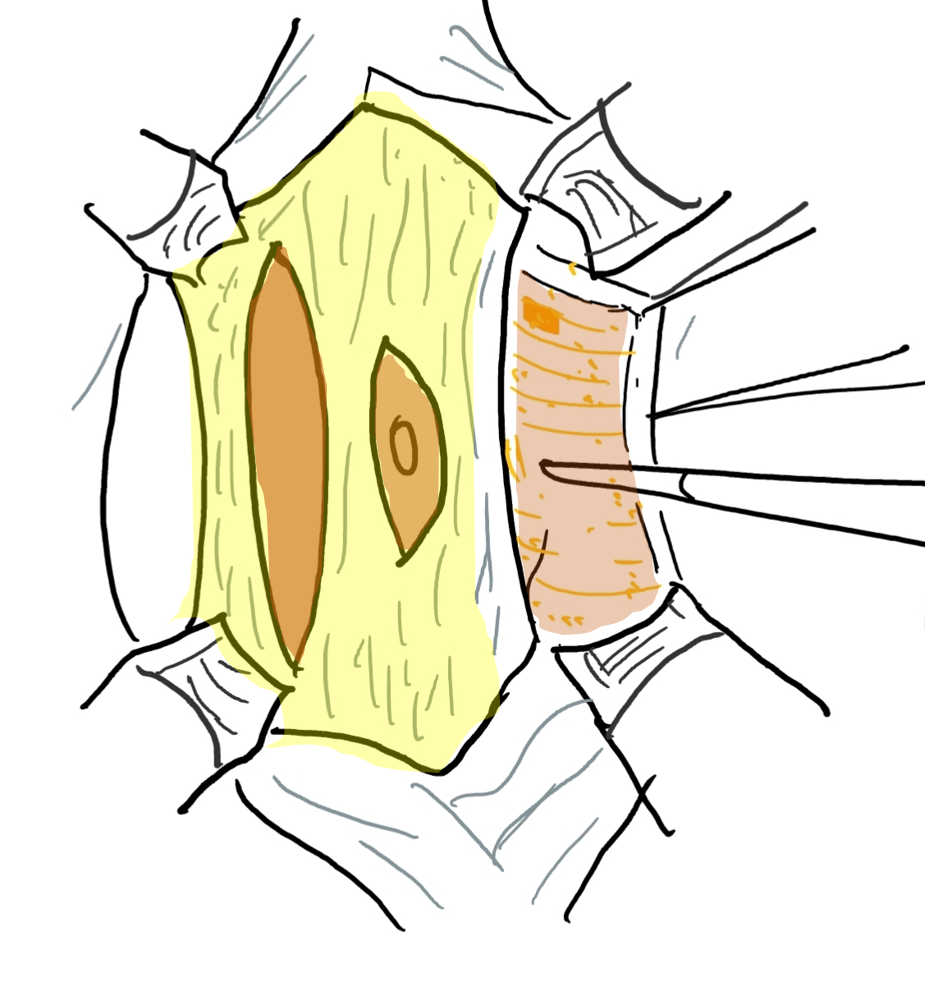
Figura 6 A camada fascial é aberta por meio de uma incisão lateral e paralela ao trajeto das artérias epigástricas superior e inferior. A extensão lateral vai até a linha axilar anterior.
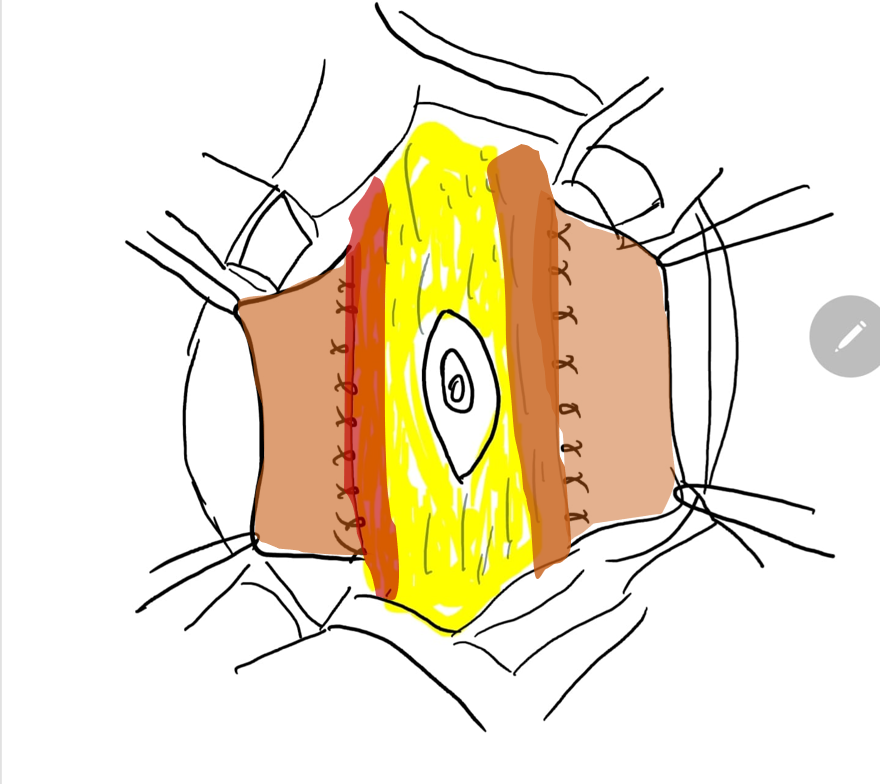
Figura 7 A margem lateral da faixa fascial central é suturada no plano profundo ao músculo.
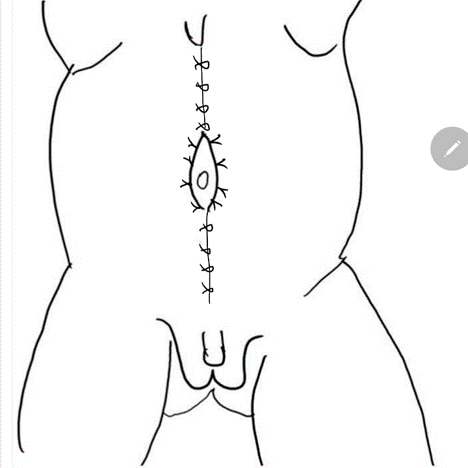
Figura 8 A fáscia lateral é então fixada na linha média acima e abaixo do umbigo. O fechamento em calça-sobre-colete fornece suporte adicional e os retalhos cutâneos de ambos os lados são fechados são fechados posteriormente.
Criptorquidia
Testículos intra-abdominais bilaterais são outra característica distintiva da PBS. A orquidopexia tradicional em estágio único e a orquidopexia de Fowler- Stephens em estágio único versus em estágios são todas opções potenciais para o manejo dos testículos, dependendo do comprimento dos vasos gonadais, e podem ser realizadas por via aberta ou laparoscópica, conforme a preferência do cirurgião.65,66,67 A parede abdominal laxa pode levar a problemas técnicos potenciais na criação e manutenção do pneumoperitônio, que podem ser minimizados pelo uso de trocartes de expansão radial e altas taxas de fluxo de gás durante a orquidopexia laparoscópica, conforme descrito por Philip et al.67,68 Deve-se enfatizar, porém, que, na ausência de qualquer problema cardiopulmonar preponderante, a orquidopexia deve ser realizada o mais precocemente possível, conforme recomendado pela atual [diretriz da AUA](https://www.auanet.org/guidelines/guidelines/cryptorchidism-guideline) e não adiada para idades mais avançadas na infância, a fim de aumentar as chances de fertilidade e melhorar a probabilidade de um procedimento em estágio único, pois a divisão dos vasos gonadais é menos provável quando a orquidopexia é realizada precocemente.
Reconstrução Abrangente
A reconstrução abrangente no paciente com síndrome de Prune Belly envolve cistoplastia de redução, ressecção do ureter distal redundante e reimplante ureteral bilateral com afunilamento.69 A reconstrução pode ser combinada com a realização de abdominoplastia e orquidopexia bilateral, geralmente antes dos dois anos de idade. Essa abordagem agressiva tem sido utilizada em muitas séries e tem mostrado bons desfechos com respeito à função renal estável ou melhorada.
Na abordagem descrita por Woodward, é feita uma incisão abdominal inferior ampla que se estende da ponta da 12ª costela, ao longo da espinha ilíaca ântero-superior até o púbis e então curva-se superiormente até a 12ª costela contralateral. Isso tende a poupar a inervação e a vascularização segmentares da parede abdominal remanescente para reconstrução. Além disso, no momento da ressecção da parede abdominal redundante, preserva-se a porção com o maior número de fibras musculares. A exposição do trato urinário proporcionada por essa incisão é excepcional. Contudo, o uso dessa técnica diminuiu devido a preocupações quanto à interrupção dos vasos epigástricos e ao tecido abdominal lateral redundante. Em vez disso, a maioria dos cirurgiões prefere a abordagem de Monfort (Figura 4), (Figura 5), (Figura 6), (Figura 7), e (Figura 8)
O peritônio posterior é incisado ao longo do comprimento dos ureteres. Os vasos espermáticos são mobilizados distalmente, tendo-se o cuidado de não comprometer as inserções peritoneais do ducto deferente caso seja necessária uma orquiopexia de Fowler-Stephens. A bexiga é aberta na linha média com eletrocautério. Reconhecem-se os óstios ureterais posicionados lateralmente e cada ureter é mobilizado. O grande comprimento dos ureteres mobilizados permite a ressecção de seu terço distal, geralmente a porção mais anormal. Realiza-se afilamento formal ou imbricação. Em seguida, os ureteres são reimplantados, utilizando-se a técnica transtrigonal ou Leadbetter-Politano. Procede-se à colocação de stents nos ureteres operados, que são exteriorizados através da bexiga e por incisões abdominais separadas.
Antes da cistoplastia de redução, os testículos são mobilizados com um amplo retalho de peritônio posterior. Em alguns casos, os testículos podem estar aderentes à bexiga aumentada e devem ser meticulosamente mobilizados para preservar a vascularização. Se a reconstrução for realizada antes dos 2 anos de idade, uma orquiopexia padrão com vasos espermáticos íntegros pode ser realizada em 80% dos casos. Caso contrário, será necessária uma orquiopexia tardia ou uma orquiopexia de Fowler-Stephens formal
A bexiga é reduzida de tamanho por meio de ressecção do remanescente uracal e da grande cúpula vesical. Uma faixa de epitélio vesical pode ser removida de uma das paredes laterais e fechada com a técnica vest-over-pants. A bexiga é drenada com um cateter de Malecot. Os testículos previamente mobilizados são trazidos através da parede abdominal distal ao nível do tubérculo púbico. Cria-se uma bolsa de dartos no escroto e cada testículo é fixado na bolsa com suturas permanentes.
A parede abdominal redundante é então mantida tensa e marcada na linha média e lateralmente nos ângulos costovertebrais para delimitar o tecido a ser ressecado. Evitar a remoção de quantidades excessivas de parede abdominal é fundamental. A segunda linha de incisão é paralela à incisão anterior. Remove-se uma grande cunha de parede abdominal de espessura total. O fechamento da parede abdominal é iniciado pela colocação de pontos-chave com fios não absorvíveis 0 ou 1–0 nas espinhas ilíacas anteriores e em cada tubérculo púbico. Esses pontos devem incluir o periósteo do osso e toda a espessura da borda tecidual proximal oposta. A tensão criada ocasionalmente permite a remoção adicional de parede abdominal redundante lateral. Cada segmento entre os pontos-chave é fechado com suturas interrompidas que aproximam toda a espessura do tecido abdominal. O tecido subcutâneo é fechado para evitar espaço morto e a pele é aproximada com sutura subcuticular absorvível.
Desfechos a Longo Prazo
Qualidade de Vida
Embora a perspectiva geral para o paciente com PBS, quanto à sobrevivência e à qualidade de vida, tenha melhorado consideravelmente, a PBS afeta profundamente a qualidade de vida relacionada à saúde nas crianças acometidas, bem como em seus cuidadores.70 Os pacientes apresentam funcionamento físico, emocional, social e escolar comprometido, e os cuidadores relatam uma qualidade de vida globalmente inferior devido aos ônus que as famílias com crianças com doenças crônicas frequentemente enfrentam. Outro estudo demonstrou que indivíduos sobreviventes com PBS apresentavam incidência significativamente alta de condições ortopédicas, gastrointestinais e cardiopulmonares que afetavam negativamente sua qualidade de vida, ao passo que o tratamento dessas condições não geniturinárias melhora suas vidas.70,71 Esses estudos enfatizam que, sendo o urologista pediátrico o principal responsável pela saúde global desses pacientes medicamente complexos, ele deve estar ciente e preparado não apenas para tratar as complicações geniturinárias tardias da PBS, mas também para avaliar e diagnosticar as comorbidades não geniturinárias que afetam diretamente seu tratamento médico e cirúrgico e sua qualidade de vida, à medida que passam pela puberdade e entram na vida adulta.
Insuficiência Renal e Transplante
Até 30% dos pacientes, em geral aqueles com função renal inicialmente prejudicada, desenvolvem insuficiência renal crônica durante a infância ou adolescência. A doença renal terminal precoce (ESRD) é considerada secundária à displasia renal, ao passo que a insuficiência renal que ocorre mais tardiamente é frequentemente atribuída ao dano parenquimatoso decorrente de infecções repetidas e ao aumento da pressão transmitida às vias urinárias superiores gerado pelo esvaziamento incompleto.72 Yalcinkaya et al relataram que a idade mediana de início da terapia de substituição renal para meninos com PBS foi de 7 anos, significativamente menor do que em pacientes do sexo masculino com outros tipos de LUTO ou displasia renal, com idade mediana no primeiro transplante de apenas 9.3 anos.73
O transplante renal é necessário para esses pacientes para assegurar crescimento e desenvolvimento normais, e o sucesso com o transplante em pacientes com PBS pode ser esperado igual ao de outros grupos pareados por idade. Desde o primeiro relato em 1976, rins de doadores cadáveres e de doadores vivos aparentados têm sido transplantados com sucesso em pacientes com PBS, com idade ao transplante variando de 8 meses a 21 anos de idade.73,74 Antes do transplante, nefroureterectomias bilaterais são geralmente realizadas em todos os casos para evitar complicações infecciosas. Recomenda-se avaliação radiográfica e urodinâmica do trato urinário inferior para garantir a ausência de obstrução e esvaziamento vesical equilibrado. O uso de cateterismo intermitente limpo para esvaziar a bexiga descompensada não é uma contraindicação ao transplante renal. Todos os pacientes devem permanecer em profilaxia antibiótica. Uma complicação incomum do transplante na síndrome de prune-belly é a torção do aloenxerto.75 A torção, com perda do enxerto resultante, foi decorrente da falta de tônus da parede abdominal. Tem sido recomendada a nefropexia para evitar essa complicação desastrosa. Em uma revisão retrospectiva de oito pacientes com síndrome de prune-belly que foram submetidos a transplante, não houve diferença estatisticamente significativa em óbitos de pacientes, sobrevida do enxerto ou função do enxerto quando comparados a controles pareados por idade. Uma análise retrospectiva mais recente de sobrevida do enxerto em 10 anos de 47% não foi estatisticamente diferente daquela de pacientes controle pareados por idade,73,74
Função Vesical
Como mencionado anteriormente, estudos urodinâmicos de pacientes com PBS geralmente mostram complacência normal, com sensibilidade e atividade contrátil diminuídas. Como consequência, alguns pacientes tendem a reter grandes volumes na bexiga, o que leva a um aumento progressivo da capacidade vesical, associado a resíduo pós-miccional significativo. A falta de suporte abdominal, a eventual associação de poliúria e o comportamento negativo de alguns pacientes em relação à micção programada geralmente aumentam essa tendência. Portanto, para prevenir maior dilatação vesical e deterioração do trato urinário superior, todos os pacientes, mesmo aqueles submetidos à reconstrução do trato urinário inferior e abdominoplastia, devem ser treinados o mais precocemente possível e constantemente incentivados a esvaziar a bexiga em horários programados, podendo associar micção dupla ou tripla e manobras de Valsalva ou de Credé.76 No caso de micção progressivamente desequilibrada, se tal programa não for viável ou for ineficaz, a cateterização vesical intermitente é mandatória, seja pela uretra ou por um conduto de Mitrofanoff. Com o crescimento e a melhora da sensação proprioceptiva, bem como com o aumento do suporte abdominal, pode-se alcançar micção eficaz. Na série de Lopes et al, 17.4% dos pacientes submetidos à reconstrução do trato urinário necessitaram cateterização intermitente pela uretra ou por um conduto de Mitrofanoff. Raramente, algumas dessas crianças readquirem micção normal com bom esvaziamento vesical.
Crescimento e Desenvolvimento Musculoesquelético
O crescimento normal pode ser esperado na maioria dos pacientes com função renal normal, embora retardo do crescimento na ausência de comprometimento renal tenha sido observado em um terço dos pacientes em uma série.34 Deformidades torácicas, como pectus excavatum, geralmente não comprometem a função pulmonar em crianças maiores e podem apresentar alguma melhora com o crescimento e o exercício. Pacientes com a síndrome podem apresentar anomalias musculoesqueléticas que prejudicam a função e a estética, como escoliose, lordose, alterações no tornozelo e no quadril, que podem necessitar de intervenção ortopédica precoce. Em um estudo recente, pacientes mais velhos e suas famílias foram informados de que a correção dos problemas musculoesqueléticos por meio de abdominoplastia e/ou cirurgias ortopédicas era a forma mais comum de os profissionais de saúde melhorarem a qualidade de vida de seus pacientes.
Função Sexual e Fertilidade
Um padrão normal de desenvolvimento sexual secundário pode ser esperado, uma vez que a produção hormonal pelos testículos é preservada, mas a função sexual pode estar prejudicada pela ejaculação retrógrada. A infertilidade primária costuma ser a regra e, quando presente, acredita-se que seja devida a uma combinação de anomalias histológicas testiculares, com espermatogênese prejudicada, defeitos estruturais dos ductos e anomalias prostáticas levando à ejaculação retrógrada.77 Contudo, a paternidade é possível com técnicas de reprodução assistida (ART), especialmente naqueles que tiveram orquidopexia precoce bem-sucedida, e há relatos de nascidos vivos normais em pacientes adultos com PBS clássica, possibilitados por técnicas de recuperação espermática e injeção intracitoplasmática de espermatozoides.78,79,80 Gravidez normal com parto vaginal assistido também foi descrita em uma paciente do sexo feminino com a síndrome.81
Conclusões
Como na maioria das anomalias congênitas complexas, a chave para o manejo da PBS é uma abordagem multidisciplinar baseada em equipe que ofereça cuidados individualizados. Dado o amplo espectro da doença, o tratamento da síndrome de prune-belly é específico para cada paciente. Quando infecção e estase comprometem o crescimento ou a função renal, recomenda-se intervenção cirúrgica agressiva para promover a drenagem. Os testículos nesses lactentes são posicionados precocemente no escroto para fins de melhora da fertilidade e isso pode ser realizado por técnicas laparoscópicas ou abertas. Devido à condição urodinâmica variável do trato urinário inferior e aos seus efeitos sobre a função renal e a infecção urinária, todos os pacientes com síndrome de prune-belly requerem vigilância urológica cuidadosa por toda a vida.
Pontos-chave
- A síndrome de Prune-Belly (PBS) inclui uma constelação de anomalias com graus variáveis de gravidade. Os três achados principais são deficiência da musculatura abdominal, testículos intra-abdominais bilaterais e trato urinário anômalo.
- O trato urinário caracteriza-se por graus variáveis de hidronefrose, displasia renal, uréteres dilatados e tortuosos, bexiga aumentada e uretra prostática dilatada/ megalouretra.
- Anomalias associadas adicionais envolvem o trato respiratório, o trato gastrointestinal, o sistema cardíaco e o sistema musculoesquelético.
- O determinante mais importante da sobrevivência a longo prazo é geralmente a gravidade da anomalia do trato urinário, em particular o grau de displasia renal.
- A hidronefrose não obstrutiva é a regra. É a infecção renal, e não a obstrução, que representa o maior risco para a função renal.
- A intervenção urológica inicial é direcionada para a drenagem vesical e o esvaziamento adequado da bexiga, a fim de evitar infecção do trato urinário (ITU) e, assim, preservar a função renal.
- O tratamento cirúrgico consiste em reconstrução do trato urinário, abdominoplastia e orquidopexia bilateral, que podem ser realizados separadamente ou em um único tempo em crianças mais novas.
- O cuidado de pacientes com PBS requer uma ampla abordagem multidisciplinar para ajudar essas crianças a prosperar, ganhar peso e estarem preparadas para cirurgia urológica, se necessário.
Referências
- Woodard S JR, E.A.. Prune belly syndrome.
- Denes FT, Lopes RI. Prune-belly syndrome. In: Partin AW, Dmochowski RR, Kavoussi LR, Peters CA, editors. Campbell wals-wein urology. 12th ed. Philadelphia: Elsevier, Inc; 2021. DOI: 10.25060/residpediatr-2018.v8n1-07.
- Grimsby GM, Harrison SM, Granberg CF, Bernstein IH, Baker LA. Impact and frequency of extra-genitourinary manifestations of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 280 281–286. DOI: 10.1016/j.jpurol.2015.06.005.
- Wong DG, Arevalo MK, Passoni NM, Iqbal NS, Jascur T, Kern AJ. Phenotypic severity scoring system and categorization for prune belly syndrome: application to a pilot cohort of 50 living patients. BJU Int 2019; 123 (1): 130–139. DOI: 10.1111/bju.14524.
- Wheatley JM, Stephens FD, Hutson JM. Prune-belly syndrome: ongoing controversies regarding pathogenesis and management. Semin Pediatr Surg 1996; 5 (2): 95–106.
- Gonzalez R, Reinberg Y, Burke B, Wells T, Vernier RL. Early bladder outlet obstruction in fetal lambs induces rental dysplasia and the prune-belly syndrome. J Pediatr Surg 1990; 25 (3): 342–345. DOI: 10.1016/0022-3468(90)90083-l.
- Ramasamy R, Haviland M, Woodard B JR, J.G.. Patterns of inheritance in familial prune belly syndrome. Urology 2005; 65 (6). DOI: 10.1016/j.urology.2004.12.050.
- Reinberg Y, Shapiro E, Manivel JC, Manley CB, Pettinato G, Gonzalez R. Prune belly syndrome in females: A triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr 1991; 118 (3). DOI: 10.1016/s0022-3476(05)82153-5.
- Balaji KC, Patil A, Townes PL, Primack W, Skare J, Hopkins T. Concordant prune belly syndrome in monozygotic twins. Urology 2000; 55 (6). DOI: 10.1016/s0090-4295(00)00452-0.
- Iqbal NS, Jascur TA, Harrison SM, Edwards AB, Smith LT, Choi ES. Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene. BMC Med Genet 2020; 21 (1). DOI: 10.1186/s12881-020-0973-x.
- Granberg CF, Harrison SM, D D. Genetic basis of prune belly syndrome: Screening for HNF1beta gene. J Urol 2012; 187 (1).
- Stephens FD, Gupta D. Pathogenesis of the prune belly syndrome. J Urol 1994; 152: 2328–2331. DOI: 10.1016/s0022-5347(17)31669-5.
- Palmer JM, Tesluk H. Ureteral pathology in the prune belly syndrome. J Urol 1974; 111 (5). DOI: 10.1016/s0022-5347(17)60050-8.
- Gearhart JP, Lee BR, Partin AW, Epstein JI, Gosling JA, Kogan BA. A quantitative histological evaluation of the dilated ureter of childhood. II: Ectopia, posterior urethral valves and the prune belly syndrome. J Urol 1995; 153 (1). DOI: 10.1097/00005392-199501000-00069.
- Snyder HM, Harrison NW, Whitfield HN, Williams I. Urodynamics in the prune belly syndrome. Br J Urol 1976; 48 (7): 663–670. DOI: 10.1111/j.1464-410x.1976.tb06716.x.
- Kinahan TJ, Churchill BM, McLorie GA, Gilmour RF, Khoury AE. The efficiency of bladder emptying in the prune belly syndrome. J Urol 1992; 148 (2 Pt 2): 600–603. DOI: 10.1016/s0022-5347(17)36665-x.
- Favorito LA, Pires RS, Gallo CM, Sampaio FJB. Study of prostate growth in prune belly syndrome and anencephalic fetuses. J Pediatr Surg 2020; 55 (10): 2221–2225. DOI: 10.1016/j.jpedsurg.2019.10.054.
- Volmar KE, Fritsch MK, Perlman EJ, Hutchins GM. Patterns of congenital lower urinary tract obstructive uropathy: Relation to abnormal prostate and bladder development and the prune belly syndrome. Pediatr Dev Pathol 2001; 4 (5). DOI: 10.1007/s10024001-0042-1.
- Deklerk DP, Scott WW. Prostatic Maldevelopment in the prune belly syndrome: A defect in prostatic stromal-epithelial interaction. J Urol 1978; 120 (3). DOI: 10.1016/s0022-5347(17)57168-2.
- Cabral B, Majidi A, Gonzalez R. Ectopic vasa deferentia in an infant with the prune belly syndrome. J Urol 1988; 94 (4).
- Kroovand RL, Al-Ansari RM, Perlmutter AD. Urethral and genital malformations in prune belly syndrome. J Urol 1982; 127 (1): 94–96. DOI: 10.1016/s0022-3468(82)80571-x.
- Berdon WE, Baker DH, Wigger HJ, Blanc WA. The radiologic and pathologic spectrum of the prune belly syndrome. The importance of urethral obstruction in prognosis. Radiol Clin North Am 1977; 15 (1).
- Hoagland MH, Hutchins GM. Obstructive lesions of the lower urinary tract in the prune belly syndrome. J Urol 1987; 138 (2). DOI: 10.1016/s0022-5347(17)43220-4.
- Volmar KE, Nguyen TC, Holcroft CJ, Blakemore KJ, Hutchins GM. Phimosis as a cause of the prune belly syndrome: Comparison to a more common pattern of proximal penile urethra obstruction. Virchows Arch 2003; 442 (2). DOI: 10.1007/s00428-002-0730-x.
- Beasley S, Bettenay F, Hutson J. The anterior urethra provides clues to the aetiology of prune belly syndrome. Pediatr Surg Int 1988; 3–3(2–3. DOI: 10.1007/bf00182775.
- Sellers BB, McNeal R, Smith RV, Griswold WR, Mendoza SA. Congenital megalourethra associated with prune belly syndrome. J Urol 1976; 116 (6). DOI: 10.1016/s0022-5347(17)59027-8.
- Uehling DT, Zadina SP, Gilbert E. Testicular histology in triad syndrome. Urology 1984; 23 (4). DOI: 10.1016/0090-4295(84)90141-9.
- Massad CA, Cohen MB, Kogan BA, Beckstead JH. Morphology and histochemistry of infant testes in the prune belly syndrome. J Urol 1991; 146 (6): 1598–1600. DOI: 10.1016/s0022-5347(17)38178-8.
- Orvis BR, Bottles K, Kogan BA. Testicular histology in fetuses with the prune belly syndrome and posterior urethral valves. J Urol 1988; 139 (2): 335–337. DOI: 10.1016/s0022-5347(17)42403-7.
- Sayre R, Stephens R, Chonko AM. Prune belly syndrome and retroperitoneal germ cell tumor. Am J Med 1986; 81 (5). DOI: 10.1016/s0022-5347(17)44221-2.
- Woodhouse CR, Ransley PG. Teratoma of the testis in the prune belly syndrome. Br J Urol 1983; 55 (5).
- Mininberg DT, Montoya F, Okada K, Galioto F, Presutti R. Subcellular muscle studies in the prune belly syndrome. J Urol 1973; 109 (3). DOI: 10.1097/00006534-197311000-00052.
- Afifi AK, Rebeiz J, Mire J, Andonian SJ, Kaloustian VM. The myopathology of the prune belly syndrome. J Neurol Sci 1972; 15 (2). DOI: 10.1016/0022-510x(72)90003-2.
- Loder RT, Guiboux JP, Bloom DA, Hensinger RN. Musculoskeletal aspects of prune-belly syndrome. Description and Pathogenesis Am J Dis Child 1992; 146 (10): 1224–1229. DOI: 10.1001/archpedi.1992.02160220110034.
- Geary DF, MacLusky IB, Churchill BM, McLorie G. A broader spectrum of abnormalities in the prune belly syndrome. J Urol 1986; 135 (2). DOI: 10.1016/s0022-5347(17)45627-8.
- Alford BA, Peoples WM, Resnick JS, L’Heureux PR. Pulmonary complications associated with the prune-belly syndrome. Radiology 1978; 129 (2). DOI: 10.1148/129.2.401.
- Wright B JR, RF N, JC P, ET S, ME S, L.E.. Gastrointestinal malformations associated with prune belly syndrome: Three cases and a review of the literature. Pediatr Pathol 1986; 5 (3–4). DOI: 10.3109/15513818609068868.
- Salihu HM, Tchuinguem G, Aliyu MH, Kouam L. Prune belly syndrome and associated malformations. A 13-year experience from a developing country. West Indian Med J 2003; 52 (4).
- Brinker MR, Palutsis RS, Sarwark JF. The orthopaedic manifestations of prune-belly (Eagle-Barrett) syndrome. J Bone Joint Surg 1995; 77 (2). DOI: 10.2106/00004623-199502000-00012.
- Cazorla E, Ruiz F, Abad A, Monleon J. Prune belly syndrome: Early antenatal diagnosis. Eur J Obstet Gynecol Reprod Biol 1997; 72 (1). DOI: 10.1016/s0301-2115(96)02664-4.
- Fitzsimons RB, Keohane C, Galvin J. Prune belly syndrome with ultrasound demonstration of reduction of megacystis in utero. Br J Radiol 1985; 58 (688). DOI: 10.1259/0007-1285-58-688-374.
- Aaronson IA. Posterior urethral valve masquerading as the prune belly syndrome. Br J Urol 1983; 55 (5). DOI: 10.1016/s0022-3468(84)80160-8.
- Tonni G, Ida V, Alessandro V, Bonasoni MP. Prune-belly syndrome: case series and review of the literature regarding early prenatal diagnosis, epidemiology, genetic factors, treatment, and prognosis. Fetal Pediatr Pathol 2013; 31 (1): 13–24. DOI: 10.3109/15513815.2012.659411.
- Yamamoto H, Nishikawa S, Hayashi T, Sagae S, Kudo R. Antenatal diagnosis of prune belly syndrome at 11 weeks of gestation. J Obstet Gynaecol Res 2001; 27 (1). DOI: 10.1111/j.1447-0756.2001.tb01213.x.
- Seidel NE, Arlen AM, Smith EA, Kirsch AJ. Clinical manifestations and management of prune-belly syndrome in a large contemporary pediatric population. Urology 2015; 85 (1). DOI: 10.1016/j.urology.2014.09.029.
- Woods AG, Brandon DH. Prune belly syndrome. A focused physical assessment. Adv Neonatal Care 2007; 7 (3): 144–145.
- Arlen AM, Nawaf C, Kirsch AJ. Prune belly syndrome: current perspectives. Pediatric Health Med Ther 2019; 10: 75–81. DOI: 10.2147/phmt.s188014.
- Rushton HG, Majd M, Chandra R, Yim D. Evaluation of 99M technetium-dimercapto-succinic acid renal scans in experimental acute pyelonephritis in piglets. J Urol 1988; 140 (5). DOI: 10.1016/s0022-5347(17)41992-6.
- Garcia-Roig ML, Grattan-Smith JD, Arlen AM, Smith EA, Kirsch AJ. Detailed evaluation of the upper urinary tract in patients with prune belly syndrome using magnetic resonance urography. J Pediatr Urol 2016; 12 (2). DOI: 10.1016/j.jpurol.2015.11.008.
- Kaplan BS. In utero intervention in prune-belly syndrome. Pediatr Nephrol 1999; 13 (2). DOI: 10.1007/s004670050581.
- Makino Y, Kobayashi H, Kyono K, Oshima K, Kawarabayashi T. Clinical results of fetal obstructive uropathy treated by vesicoamniotic shunting. Urology 2000; 55 (1). DOI: 10.1016/s0090-4295(99)00403-3.
- Burbige KA, Amodio J, Berdon WE, Hensle TW, Blanc W, Lattimer JK. Prune belly syndrome: 35 years of experience. J Urol 1987; 137 (1). DOI: 10.1016/s0022-5347(17)43880-8.
- Fallat ME, Skoog SJ, Belman AB, Eng G, Randolph JG. The prune belly syndrome: A comprehensive approach to management. J Urol 1989; 142 (3). DOI: 10.1016/s0022-5347(17)38895-x.
- Tank ES, McCoy G. Limited surgical intervention in the prune belly syndrome. J Pediatr Surg 1983; 18 (6). DOI: 10.1016/s0022-3468(83)80004-9.
- Woodhouse CRJ, Kellett MJ, Williams DI. Minimal surgical interference in the prune belly syndrome. Br J Urol 1979; 51 (6). DOI: 10.1111/j.1464-410x.1979.tb03582.x.
- Woodard P JR, T.S.. Reconstruction of the urinary tract in prune belly uropathy. J Urol 1978; 119 (6). DOI: 10.1016/s0022-5347(17)57644-2.
- Bukowski TP, Perlmutter AD. Reduction cystoplasty in the prune belly syndrome: a long-term followup. J Urol 1994; 152 (6 Pt 1): 2113–2116. DOI: 10.1016/s0022-5347(17)32333-9.
- Wille MA, Jayram G, Gundeti MS. Feasibility and early outcomes of robotic-assisted laparoscopic Mitrofanoff appendicovesicostomy in patients with prune belly syndrome. BJU Int 2011; 109 (1). DOI: 10.1111/j.1464-410x.2011.10317.x.
- Ehrlich RM, Lesavoy MA, Fine RN. Total abdominal wall reconstruction in the prune belly syndrome. J Urol 1986; 136: 282–285. DOI: 10.1016/s0022-5347(17)44842-7.
- Randolph J, Cavett C, Eng G. Abdominal wall reconstruction in the prune belly syndrome. J Pediatr Surg 1981; 16 (6): 960–964. DOI: 10.1016/s0022-5347(17)52848-7.
- Monfort G, Guys JM, Bocciardi A, Coquet M, Chevallier D. A novel technique for reconstruction of the abdominal wall in the prune belly syndrome. J Urol 1991; 146 (2 Pt 2): 639–640. DOI: 10.1016/s0022-5347(17)37880-1.
- Denes FT, Lopes RI, Oliveira LM, Tavares A, Srougi M. Modified abdominoplasty for patients with the Prune Belly syndrome. Urology 2014; 83 (2): 451–454. DOI: 10.1016/j.urology.2013.09.031.
- Fearon JA, Varkarakis G. Dynamic abdominoplasty for the treatment of prune belly syndrome. Plast Reconstr Surg 2012; 130 (3). DOI: 10.1097/prs.0b013e31825dc170.
- Franco I. Laparoscopic assisted modification of the firlit abdominal wall plication. J Urol 2005; 174 (1). DOI: 10.1097/01.ju.0000161214.65458.c2.
- Patil KK, Duffy PG, Woodhouse CR, Ransley PG. Long-term outcome of Fowler-Stephens orchiopexy in boys with prune belly syndrome. J Urol 2004; 171 (4): 1666–1669. DOI: 10.1097/01.ju.0000118139.28229.f5.
- Yu T-J, Lai M-K, Chen W-F, Wan Y-L. Two-stage orchiopexy with laparoscopic clip ligation of the spermatic vessels in prune-belly syndrome. J Pediatr Surg 1995; 30 (6). DOI: 10.1016/0022-3468(95)90768-8.
- Philip J, Mullassery D, Craigie RJ, Manikandan R, Kenny SE. Laparoscopic orchidopexy in boys with prune belly syndrome—Outcome and technical considerations. J Endourol 2011; 25 (7). DOI: 10.1089/end.2010.0257.
- Saxena AK, Brinkmann OA. Unique features of prune belly syndrome in laparoscopic surgery. J Am Coll Surg 2007; 205 (2). DOI: 10.1016/j.jamcollsurg.2007.03.008.
- Lopes RI, Tavares A, Srougi M, Dénes FT. 27 years of experience with the comprehensive surgical treatment of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 276 271–277. DOI: 10.1016/j.jpurol.2015.05.018.
- Arlen AM, Kirsch SS, Seidel NE, Garcia-Roig M, Smith EA, Kirsch AJ. Health-related quality of life in children with Prune belly Syndrome and their caregivers. Urology 2016; 87 (224). DOI: 10.1016/j.urology.2015.09.028.
- Lesavoy MA, Chang EI, Suliman A, Taylor J, Kim SE, Ehrlich RM. Long-term follow-up of total abdominal wall reconstruction for prune belly syndrome. Plast Reconstr Surg 2012; 129 (1). DOI: 10.1097/prs.0b013e3182362091.
- Reinberg Y, Manivel JC, Pettinato G, Gonzalez R. Development of renal failure in children with the prune belly syndrome. J Urol 1991; 145 (5): 1017–1019. DOI: 10.1016/0022-3468(92)90159-5.
- Alcinkaya F, Bonthuis M, Erdogan BD, Stralen KJ, Baiko S, Chehade H, et al.. Outcomes of renal replacement therapy in boys with prune belly syndrome: findings from the ESPN/ERA-EDTA Registry. Pediatr Nephrol 2018; 33 (1): 117–124. DOI: 10.1007/s00467-017-3770-9.
- Fontaine E, Salomon L, Gagnadoux MF, Niaudet P, Broyer M, Beurton D. Long-term results of renal transplantation in children with the prune-belly syndrome. J 1997; Urol.158(3 Pt 1):892-4. DOI: 10.1097/00005392-199709000-00067.
- Marvin RG, Halff GA, Elshihabi I. Renal allograft torsion associated with prune-belly syndrome. Pediatr Nephrol 1995; 9 (1). DOI: 10.1007/bf00858981.
- Smith CA, Smith EA, Parrott TS, Broecker BH, Woodard JR. Voiding function in patients with the prune-belly syndrome after Monfort abdominoplasty. J Urol 1998; 159 (5). DOI: 10.1016/s0022-3468(99)90298-1.
- Woodhouse CR, Snyder HM. Testicular and sexual function in adults with prune belly syndrome. J Urol 1985; 133 (4): 607 9. DOI: 10.1016/s0022-5347(17)49108-7.
- Halpern JA, Das A, Brannigan RE. Successful sperm retrieval in prune belly syndrome. Asian J Urol 2020; 7 (4): 376–378. DOI: 10.1016/j.ajur.2019.07.004.
- Kolettis PN, Ross JH, Kay R, Thomas AJ. Sperm retrieval and intracytoplasmic sperm injection in patients with prune-belly syndrome. Fertil Steril 1999; 72 (5). DOI: 10.1016/s0015-0282(99)00388-x.
- Fleming SD, Varughese E, Hua VK, Robertson A, Dalzell F, Boothroyd CV. Normal live births after intracytoplasmic sperm injection in a man with the rare condition of Eagle-Barrett syndrome (prune-belly syndrome. Fertil Steril 2013; 100 (6). DOI: 10.1016/j.fertnstert.2013.07.1994.
- Hillman RT, Garabedian MJ, Wallerstein RJ. Pregnancy outcome in a woman with prune belly syndrome. BMJ Case Reports 2012. DOI: 10.1136/bcr-2012-006490.
Ultima atualização: 2025-09-21 13:35