
27: 无腹肌综合征
阅读本章大约需要 11 分钟。
引言
梅干腹综合征(PBS),又称 Eagle-Barrett 综合征,是一种罕见的多系统性疾病,典型特征为一组严重程度不一的异常,主要表现为腹壁肌发育不全、双侧腹腔内睾丸以及泌尿道异常。1,2此外,相关的非泌尿生殖系统异常可累及呼吸道、胃肠道、心脏系统和肌肉骨骼系统。3 疾病严重程度呈广泛连续谱,部分患儿未能存活过新生儿期,亦有患者仅轻度受累。4
胚胎学
关于PBS的胚胎发生,已经提出了几种主要理论,但尚无一种获得普遍认可,而且它们之间存在一定的重叠。四种主要理论为:1,宫内早期发生后尿道梗阻(可能为前列腺发育不全/发育异常或异常的尿道),导致近端膀胱、输尿管和肾脏的扩张,并伴随腹壁发育不良;2,侧板中胚层的原发性缺陷,其为输尿管、膀胱、前列腺、尿道和睾丸引带的前体;3,泌尿道的内在缺陷,导致输尿管扩张和胎儿腹水;以及4,卵黄囊缺陷。5,6
流行病学
据报道,PBS 的发病率为每 29,000 至每 40,000 例活产中有 1 例,与膀胱外翻的发病率相似,其中 95% 的受累患者为男性。1,2,7 女性占所有 PBS 病例的不到 5%,其表现为腹壁缺陷及扩张、畸形的尿路,且无相关的性腺异常。8 在双胎、黑人群体以及由较年轻的母亲所生儿童中发病率更高。9 尽管 PBS 通常呈散发发生,但也怀疑存在遗传学因素参与,与编码平滑肌转录因子、收缩肌丝以及神经酶/受体的基因突变有关。10,11
临床表现与发病机制
泌尿生殖系统畸形
肾脏
PBS患者的肾脏异常谱从正常肾实质到发育不良不等。约有一半病例存在肾发育不良,通常属于Potter II型和IV型,而且双侧之间也可不同。1,2 其中,II型由于肾单位稀少且肾实质结构紊乱,更提示为肾间质缺陷;而IV型伴有皮质及小管囊肿,则与出口梗阻相关,且未通过尿囊管减压。12 虽然肾集合系统具有特征性扩张,且常常达到严重程度,但扩张程度与肾发育不良的严重程度并不相关,即便在巨大扩张的情况下,肾盏形态也可能保存良好。尽管肾积水通常为非梗阻性,仍可出现原发性或继发性肾盂输尿管连接部梗阻。然而,需要注意的是,在这些患者中,相较于梗阻,肾脏感染才是对肾功能构成最大风险的因素。1,2
输尿管
输尿管通常表现为扩张、迂曲且冗长。尽管显著的扩张与狭窄可发生于各个水平,但输尿管的近端(上段)通常较远端节段异常程度轻。膀胱输尿管反流(VUR)在75%的PBS患儿中存在。12 梗阻并不常见,但已在肾盂输尿管连接部和输尿管膀胱连接部有所报道。组织学切片显示,在受累较重的远端节段,尤其是发生反流的节段,平滑肌细胞缺乏,且胶原与平滑肌的比例增加;而近端节段则可见外观更为正常的平滑肌细胞。13 在进行输尿管重建时,这一点至关重要。超微结构检查所见的粗、细肌原纤维数量减少,被认为由于输尿管壁对合不良而导致蠕动无效,进而使上尿路出现淤滞,这可能引发感染。此外,在许多患者中,尿路异常的严重程度并不与腹壁松弛的程度成正比。14
膀胱
膀胱通常显著增大,脐尿管处可见假性憩室,且在25%~30%的患者中可见脐尿管通畅。12 与由其他原因导致梗阻的膀胱所见不同,尽管膀胱壁非常增厚,其轮廓仍平滑。排尿时,膀胱颈广泛开放,通向扩张的前列腺部尿道。1,2 组织学上,在无梗阻时,膀胱中胶原与肌纤维的比值增高;然而当存在梗阻时可见平滑肌肥大。膀胱镜检查可见三角区被牵开,输尿管开口向上、向外移位,这可能促成膀胱输尿管返流的高发生率。尿动力学评估的特征性发现包括膀胱顺应性正常、首次排尿感觉延迟以及容量大。15 这些膀胱的排尿效率不一,有些可完全排空,另一些则存在显著的排尿后残余,可能与相对性出口梗阻以及这些患者的逼尿肌在排尿时无法产生足够压力有关。16 术语”非平衡”排尿通常用于指代前一种情况,即相对的流出道阻力阻止了有效的膀胱排空。1,2 尽管50%的梅干腹患者能自发排尿,且排尿压力、尿流率正常、排尿后残余量低,但已观察到,随生长发育,平衡排尿可能出现恶化,导致显著的排尿后残余,强调了定期评估的必要性。16
前列腺及附属性腺
后尿道扩张是由于前列腺发育不全,可能与异常的间充质-上皮发育有关。12,13,14,15,16,17,18,19 组织学切片显示前列腺细胞成分很少,上皮细胞和平滑肌细胞减少,而结缔组织细胞增多。19 大约20%的患者在后尿道远端也观察到梗阻性病变,表现为尿道闭锁、尿道瓣膜、尿道狭窄、尿道隔膜和尿道憩室。 前列腺实质组织的缺乏可导致排尿时尿道成角,Stephens将其称为IV型瓣膜。12 前列腺发育不全及其相关的膀胱颈关闭不全也被认为是PBS患者出现射精失败/逆行射精的因素之一。1,2 输精管和精囊等附属性腺常见闭锁,但二者中的任一者亦可表现为扩张或增厚。 附睾可能与睾丸附着不良,并且输出小管与睾网之间也可能缺乏连续性。20
前尿道
尽管 PBS 患儿的前尿道通常正常,仍有关于该尿道段的若干异常被报道;其中最常见的是尿道闭锁或发育不全以及巨尿道。21,22,23,24,25 有人推测,尿道闭锁或发育不全的发生是由于尿道未被使用,而非畸形。若未伴有通过脐尿管未闭进行的减压,或有时甚至因膀胱自发性破裂并形成瘘管而减压,尿道闭锁往往是致命的。22
PBS 的一个独特特征是出现巨尿道。26 已有观点提出,宫内龟头部尿道与阴茎尿道交界处的短暂性阻塞可能导致巨尿道。在 PBS 患者中可见两种巨尿道变异,即梭形型(图 1)和舟形型(图 2),(图 3)类型。24 梭形型为阴茎海绵体以及尿道海绵体的发育不全,可能源自尿道褶皱间充质的缺陷,临床上表现为排尿时整个阴茎的扩张。另一方面,舟形型可能起因于尿道支持组织的间充质缺陷,仅致尿道海绵体发育不全,而龟头与阴茎海绵体(corpora cavernosa)得以保留,其特征为排尿时腹侧尿道扩张。26
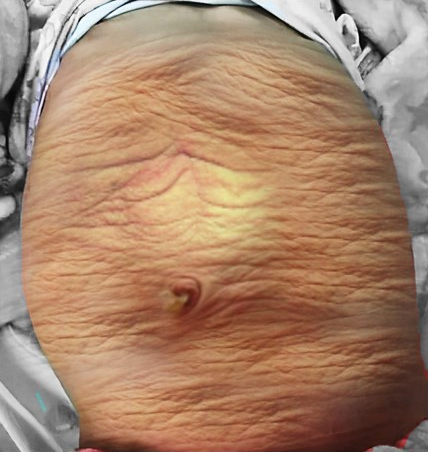
图 1 患有梅干腹综合征的儿童的临床照片
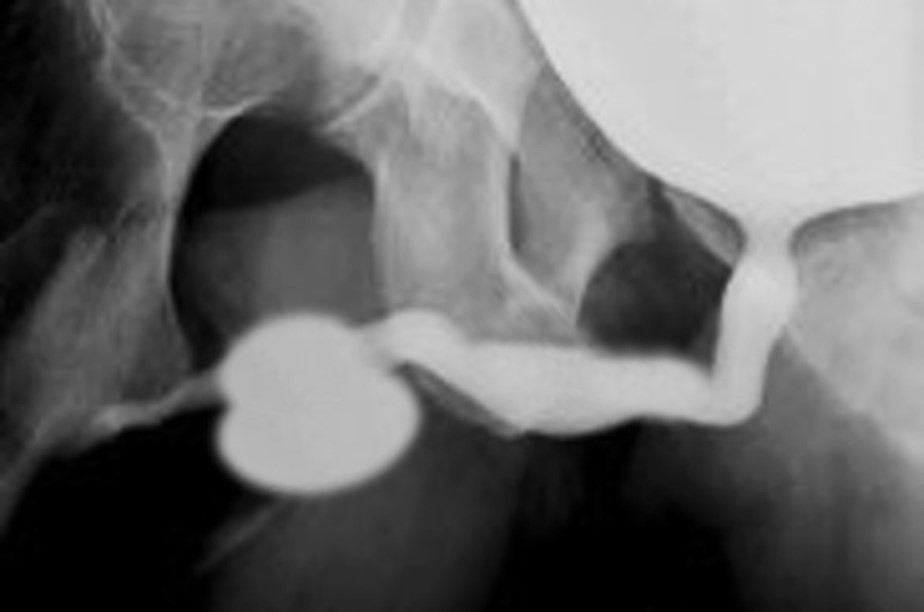
图 2 梭形巨尿道
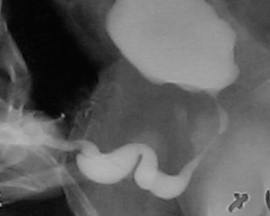
图 3 舟状型巨尿道
睾丸
位于髂血管之上并邻近扩张的输尿管的双侧腹内睾丸被认为是PBS的典型发现之一。1,2 尽管长期以来,人们将巨膀胱导致的机械性梗阻以及因腹壁异常造成的腹内压不足等机械力假设为睾丸下降失败的病因,但有些具有典型泌尿道与腹壁肌肉异常的患者却可见睾丸已下降,这一事实使人们对单纯机械因素产生怀疑。1,2 关于PBS患儿的睾丸是否与非PBS者存在内在差异,Pak et al 比较了腹内睾丸的组织学,发现PBS睾丸与非PBS胎儿腹内睾丸在生殖细胞数量、精原细胞和莱迪希细胞方面无差异。27,28 然而,Orvis et al 指出,胎儿PBS睾丸中精原细胞数量减少且莱迪希细胞增生,提示存在内在性睾丸异常。29 关于这些睾丸的恶性肿瘤风险,尽管由于缺乏生精上皮其风险可能相对较低,但仍需尽早行睾丸固定术并进行长期随访,以降低睾丸恶性肿瘤的风险并提高检出率。30,31
腹壁缺陷
PBS 患儿的腹壁外观具有特征性。最常见的是肌肉缺如呈不均匀、斑片状分布,内侧和下部肌肉节段通常最为缺如,且其程度可能与泌尿道异常不成比例。受累最严重的区域可能仅有皮肤、皮下脂肪以及腹膜表面的单层纤维层,采用电子显微镜的组织学检查显示非特异性的肌丝排列紊乱、Z 线紊乱及线粒体增生。32,33 尽管存在这些腹壁问题,这些儿童的伤口愈合良好,且无感染或切口疝的倾向。出生时的典型外观为皮肤松弛多皱,腹部在侧腹部膨隆。随着患儿生长,其中一些人的腹部张力和外观改善、皱褶减少,而另一些人则更呈鼓肚样大腹外观。1,2 步态通常不受影响,尽管行走发育可能延迟;这些儿童倾向于先翻向侧卧位,并借助手臂从仰卧位坐起。下胸壁支持不良可导致肋缘外翻,由此咳嗽效能受限,使这些儿童更易罹患呼吸道疾病。
泌尿生殖系统外异常
在所有PBS患儿中,75%存在非泌尿道异常。1,2,4 除了明显的腹壁缺损之外,最常见的问题为心脏、肺部和骨科(表 1)。34,35,36,37,38,39,40 除这些器官特异性疾病之外,近50%的PBS患儿为早产,这显著增加了合并症的发生。
表 1 在无腹肌综合征中见到的常见泌尿生殖系统外畸形
| 系统 | 常见缺陷 | 备注 |
|---|---|---|
| 心脏异常 (10%) | 动脉导管未闭、房间隔和室间隔缺损、室间隔缺损、法洛四联症 | 出生时的心脏异常可能优先于泌尿系统问题而处理。 |
| 肺部异常 (55%) | 肺发育不全 气胸和纵隔气肿可伴或不伴肺发育不全出现 | 与肾发育不良相关的重度少羊水或严重的膀胱出口梗阻可导致肺发育不全,并可能导致新生儿死亡。近一半PBS新生儿需要气管插管和机械通气支持,伴随相应的并发症。由于无法产生显著的腹腔内压力以及相关的肌肉骨骼异常,如脊柱侧弯和胸廓异常(最常见为漏斗胸),这些儿童发生肺炎和肺叶不张的风险增加。 |
| 胃肠道异常 (30%) | 这些异常源于中肠旋转不全,形成宽系膜,从而使肠道活动度增加,可见肠旋转不良、肠扭转、闭锁和狭窄。亦有报道因系膜固定异常导致的脾扭转。亦有脐膨出、腹裂、腹部内脏反位、VACTERL(椎体畸形、肛门闭锁、心脏缺陷、气管食管瘘、肾脏异常和肢体畸形)以及肛门直肠异常的报道。 | 由于腹壁肌发育不全导致产生腹腔内压力的能力受限,便秘成为终身性问题,并可能导致获得性巨结肠。 |
| 骨科异常 (65%) | 膝关节外侧的酒窝样凹陷是少羊水的常见发现,在PBS患者中亦常见,并常与其他缺陷同时出现,如马蹄内翻足(26%)、髋关节发育不良(5%)和先天性脊柱侧弯(4%)。下肢发育不全、缺如或截肢。 | 鉴于这些肌肉骨骼缺陷多为单侧,最可能的病因是少羊水的压迫效应。另一种提出的下肢畸形机制是膨胀的膀胱压迫髂外血管,从而影响下肢的血供。此外,其中一些畸形如肾性骨营养不良、髋关节脱位、脊柱侧弯以及漏斗胸或鸡胸,随着生长而出现或加重。 |
诊断与评估
产前
尽管已有报道显示在妊娠11–12周即可通过超声诊断 PBS,但由于 PBS 在产前的许多超声所见与由其他原因导致的下尿路梗阻(LUTO)相似(包括后尿道瓣膜、输尿管囊肿和尿道闭锁),其诊断准确率可从30%到85%不等。40,41,42,43,44 每当能清晰识别以下超声异常时,应考虑 PBS 的产前诊断:羊水过少、泌尿系统异常(泌尿道扩张、巨大膀胱、双侧输尿管肾积水)以及腹壁肌肉缺如。早期诊断不仅可通过促使经治医师安排在三级医疗中心分娩,以便对受累新生儿进行及时的多学科管理而提高生存率,也可在准父母有此意愿时提供自愿终止妊娠的选择。43
新生儿
在新生儿中,腹壁的外观具有诊断意义,无论产前是否已知诊断。患有 PBS 的患儿提出独特挑战,并需要由多学科团队及时管理,该团队包括新生儿科医师、泌尿科医师、肾脏科医师,并在需要时纳入心脏科医师和小儿骨科医师。鉴于疾病严重程度差异很大,必须在新生儿期进行多系统评估,以排除重要的心脏、肺部或其他相关畸形。应立即进行胸部X线检查,以排除合并的气胸和纵隔气肿。与 LUTO 一样,出生后初期病程取决于合并症的严重程度(如肺发育不全);在早产和严重肺发育不全者中预后更差。45,46,47
这些婴儿的孕期羊水过少提示肾功能受损。出生时的初始肌酐测定反映母体肾功能,因此需要反复取样。如果在出生后48–72小时,足月儿的血清肌酐大于1.0或早产儿大于1.5,则提示存在一定程度的肾功能不全。生命最初几周内血清肌酐进行性升高预示预后不良。如果初始最低肌酐值小于0.7 mg/dL,则随后发生肾功能衰竭的可能性不大。48 送尿做基线培养,并开始抗生素预防性治疗。出生时测定血清和尿液电解质有助于评估钠的保留能力,提示肾功能良好。
诊断评估
对于因其他合并症而需先行病情稳定的患者,应在病情稳定后进行全面的泌尿系统评估。初始应进行体格检查和泌尿道超声检查。46 肾脏超声可提供肾皮质厚度、是否存在囊性改变以及肾脏大小等信息。排尿前后进行超声检查可提示是否存在膀胱输尿管反流及排尿后残余尿。
起初,对于肾功能异常的婴儿会进行排尿性膀胱尿道造影(VCUG),以确保不存在真正的解剖性尿道梗阻。必须识别这种“致命变异”,以及将后尿道瓣膜“伪装”成梅干腹综合征的潜在混淆,因为治疗会发生巨大变化。此外,VCUG可在多达85%的患者中显示膀胱输尿管反流,并评估膀胱排空。所有婴儿在进行无菌导尿前均接受抗生素治疗,以降低感染风险。总体而言,泌尿道的影像学评估会根据个体泌尿外科医师的管理理念进行个体化制定。对于肾功能良好的婴儿,应避免进行需要高风险器械操作的造影检查。22
在初步评估之后,巯基乙酰三甘氨酸(MAG3)肾显像可同时提供功能和解剖学信息。可以评估血流、分肾功能以及对呋塞米(Lasix)后引流的反应,并与后续检查进行比较。已注意到利尿性肾图在显示无腹肌综合征患者梗阻方面的局限性。49 有时需要进行 Whitaker 试验作为确诊性检查以证明存在显著梗阻,尽管这主要具有历史意义。进行二巯基丁二酸(DMSA)肾显像而非静脉尿路造影(IVP),以评估肾脏大小、管状质量和分肾功能。由于这些患者需要长期随访,比较性研究对于评估肾脏生长和瘢痕极为宝贵。一些团队主张在这些患者中采用磁共振尿路成像(MRU),因为其具有高分辨率并能提供优异的功能和解剖学数据,从而可对上尿路异常进行细致可视化,这也可能有助于手术规划。50 如表 1所示,Woodard 描述的新生儿表现可分为三大类别。1,2
治疗与结局
产前管理
宫内干预
随着母胎先进影像与诊断技术的改进,妊娠早期即可通过影像学怀疑皱腹综合征。胎儿干预通常针对与肺发育不全相关的羊水过少或无羊水,而肺发育不全是围产期死亡的首要原因。宫内干预的标准包括妊娠第二或第三孕期、重度羊水过少、巨大膀胱、重度肾积水、核型正常、其他已检出先天畸形的范围有限,以及系列胎儿抽吸尿液分析结果良好。51,52,53 尽管已有多项关于用于胎儿梗阻性尿路病变的膀胱-羊膜腔分流术的报告,综述未能证实胎儿干预对随后肾功能具有有益影响,且即便恢复至正常羊水量,也无法保证肺功能。由于大多数皱腹综合征患儿无可证实的梗阻证据,且肾功能与泌尿道扩张程度不相关,必须谨慎权衡宫内干预的风险与获益。53
出生后管理
管理的总体目标是保护肾功能并预防感染。实现这些目标可以通过多种治疗方法,从密切观察随访到立即进行尿路外科重建。新生儿期一般避免早期外科干预,除非出现肌酐升高或发生感染,需要尽早行膀胱造瘘术。如表2所示,这些患者的管理取决于初诊时的严重程度分级。
表 2 根据 Woodwards 的新生儿期表现标准划分的不同类别之临床表现与处理
| 严重程度类别 | 临床表现 | 处理 | 随访与结局 |
|---|---|---|---|
| I | 明显的羊水过少,继发于肾发育不良和/或出口梗阻,导致严重的肺发育不全和骨骼异常 | 在少数幸存于肺部损伤的婴儿中,曾尝试通过皮肤肾盂造口进行高位尿路分流;然而,由于存在严重的基础性肾发育不良,通常无法恢复肾功能。 | 这些新生儿常在数日内死亡,可进行的干预有限。与产前羊水过少及产后肺发育不全相关者,通常在生后即刻期因肺部并发症而死亡。 |
| 可出现 Potter 面容,为羊水过少的继发表现。特征包括小下颌、眼距增宽、睑裂扁平、内眦赘皮明显、鼻梁低平、低位耳且缺乏软骨,以及骨骼畸形。 | 仅需导尿即可 | ||
| 尿道闭锁的病例通常属于这一最为严重的类别。 | |||
| II | 中度肾功能不全和中-重度输尿管肾盂积水,肺发育不全并非突出特征。 | 这些儿童给予长期预防性抗生素,并定期监测感染及任何肾功能下降。 | 需要终身术后监测,通过功能和解剖学检查评估肾功能以及上下尿路的最佳引流。 |
| 在有感染证据、肾脏生长不足或肾功能下降的情况下,积极进行尿路重建。 | |||
| 然而,手术重建通常推迟至婴儿满3个月或更晚,以允许肺成熟。 | |||
| 若肾功能不全严重或起初就存在感染,建议通过皮肤膀胱造口进行一段时间的尿路分流。 | |||
| 皮肤输尿管造口术可能对改善引流帮助有限。此外,上段输尿管在解剖学和组织学上最为接近正常,若日后选择个体化重建,应予以保留。 | |||
| III | 轻度三联征表现或不完全形式 | 占多数患者;尽管上下尿路明显扩张,仍表现出良好的肾功能。 | 类似于第2类,需要终身监测。 |
| 肾功能通常正常或轻度受损,且无肺功能不全。 | 通常不需要尽早进行尿路重建手术,这些患者通过长期抗生素预防以及采用选择性影像学技术对肾功能进行序贯评估来管理。 | 一旦出现泌尿道感染或肾功能下降,应进一步进行特异性评估,如尿动力学检查或利尿肾动态显像,以证实可能需要干预的下尿路或上尿路梗阻。 | |
| 这些患者可从早期睾丸固定术和腹壁成形术中获益,因为纠正腹壁可提高排便和排尿效率。 |
梅干腹综合征的手术干预
泌尿道
在 PBS 中的一个特征性发现是全泌尿道的低压扩张,其范围从近端的肾盂延伸至远端的尿道。膀胱通常增大且低张,顺应性增高,约有75%的患者存在低压膀胱输尿管返流 (VUR)。虽然这些大容量、顺应性良好的膀胱可实现低压储尿,但由于逼尿肌收缩力降低,常表现为排空不全。因此,初始泌尿科干预的重点是膀胱引流,以避免泌尿道感染 (UTI),从而保护肾功能。为此,常实施包皮环切术、预防性使用抗生素以及定时/双重排尿,以降低 UTI 的发生率并保护上尿路免受进一步损伤.54,55,56,57 当此类保守治疗未能预防感染并实现充分排空时,这些患儿可能需要减容膀胱成形术和 /或 阑尾膀胱造口术,以便进行清洁间歇导尿,和/或进行抗返流手术以处理相关的 VUR.58,59,60
腹壁
将较为正常的周围腹壁前移以支持该异常且缺损的中央部分的腹壁重建,是 PBS 患者外科治疗中的重要考量。除外观美观性外,已证明腹壁成形术还能在不依赖泌尿生殖道重建的情况下改善膀胱动力学功能。术前常进行尿动力学检查,以确定是否需要任何伴行的膀胱手术,例如在排空不全时行阑尾膀胱造口术,和/或在存在膀胱输尿管返流(VUR)时行输尿管再植术。传统上,文献中报道了三种用于矫正与 PBS 相关腹壁缺损的手术及其改良。61,62,63 Randolph 手术通过切除下腹壁的一部分以矫正筋膜的纵向冗余,而 Monfort (图 4), (图 5), (图 6), (图 7), (图 8) 和 Ehrlich 手术均采用筋膜的纵向重叠来矫正侧向冗余,同时增强腹壁的强度。亦有腹腔镜入路的腹壁成形术之报道,证明其既有助于重建本身,也有助于界定肌肉缺损的不同程度。64 还需注意,PBS 患者在腹壁肌肉缺损的严重程度和分布模式方面可能各具特征,例如某一特定区域完全缺乏肌肉。Smith 及其同事近期报告了他们对 Monfort 手术的改良,使之能够更个体化地矫正腹壁松弛,并将脐的位置调整到更符合解剖学的部位。
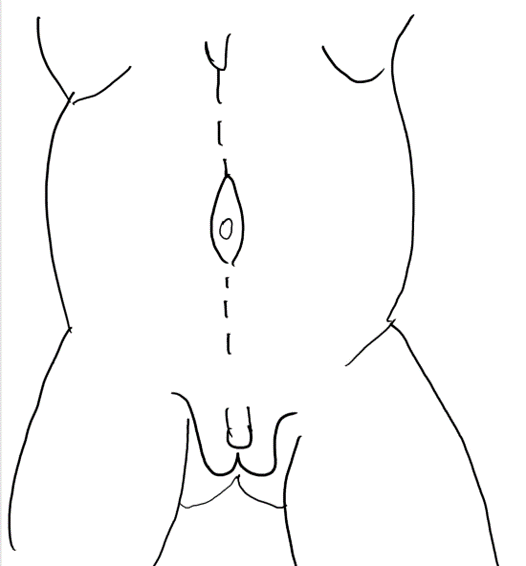
图 4 自剑突至耻骨的正中切口,绕行脐部。
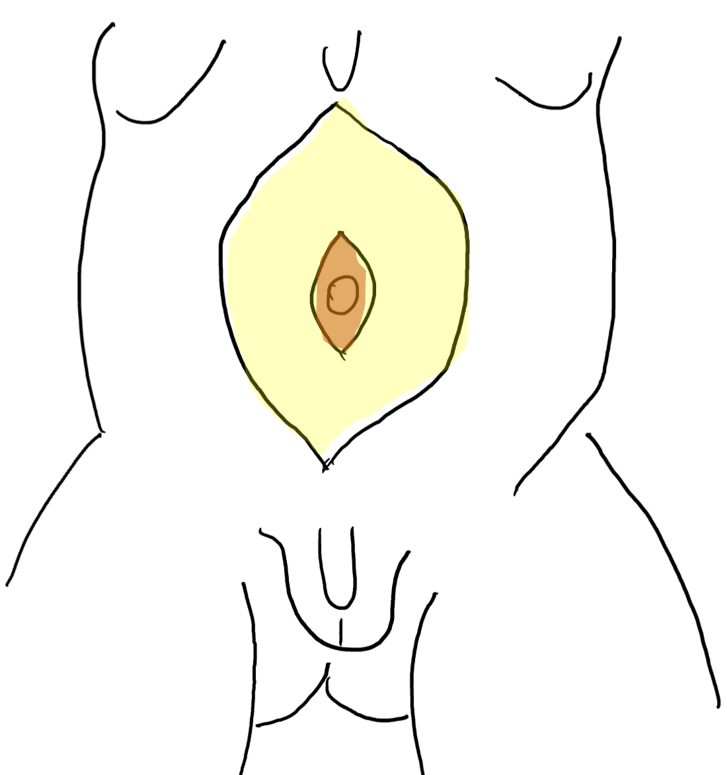
图 5 在皮下脂肪与筋膜层之间掀起皮瓣。脐由中央筋膜条带支撑。
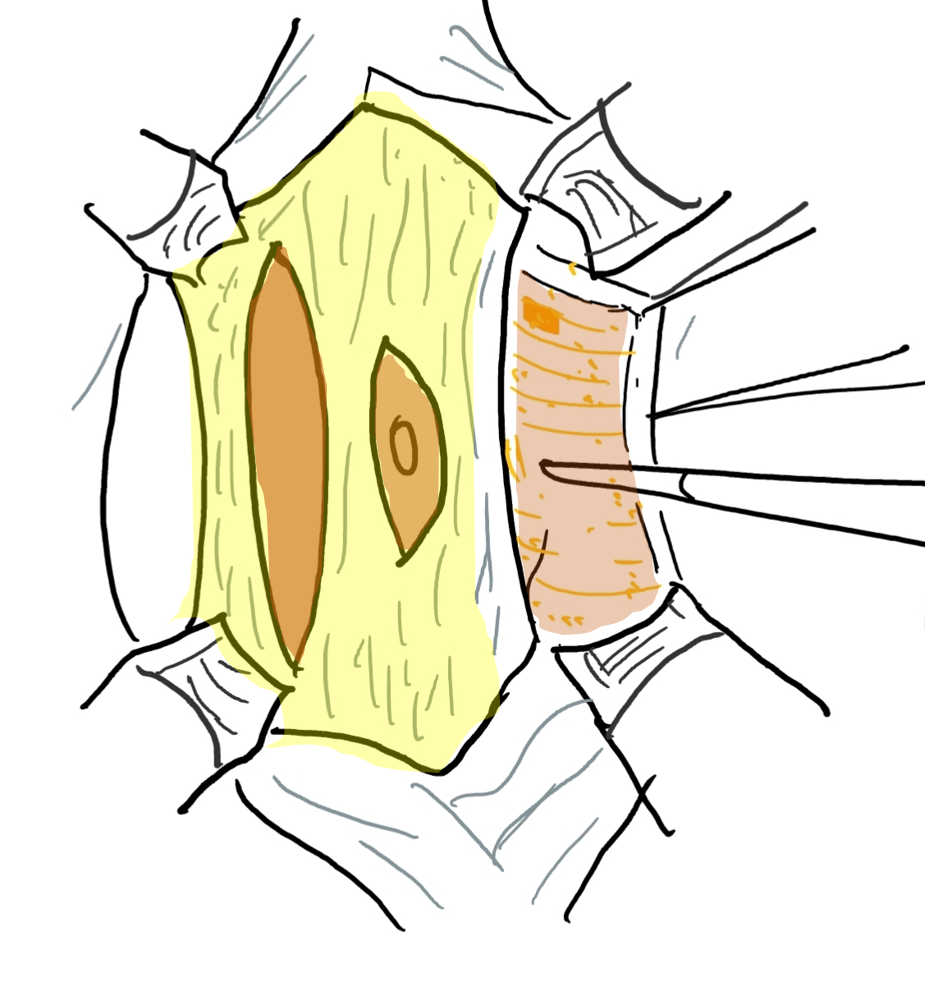
图6 筋膜层通过在上、下腹壁动脉走行的外侧且与其平行的切口予以切开。外侧延伸达前腋线。
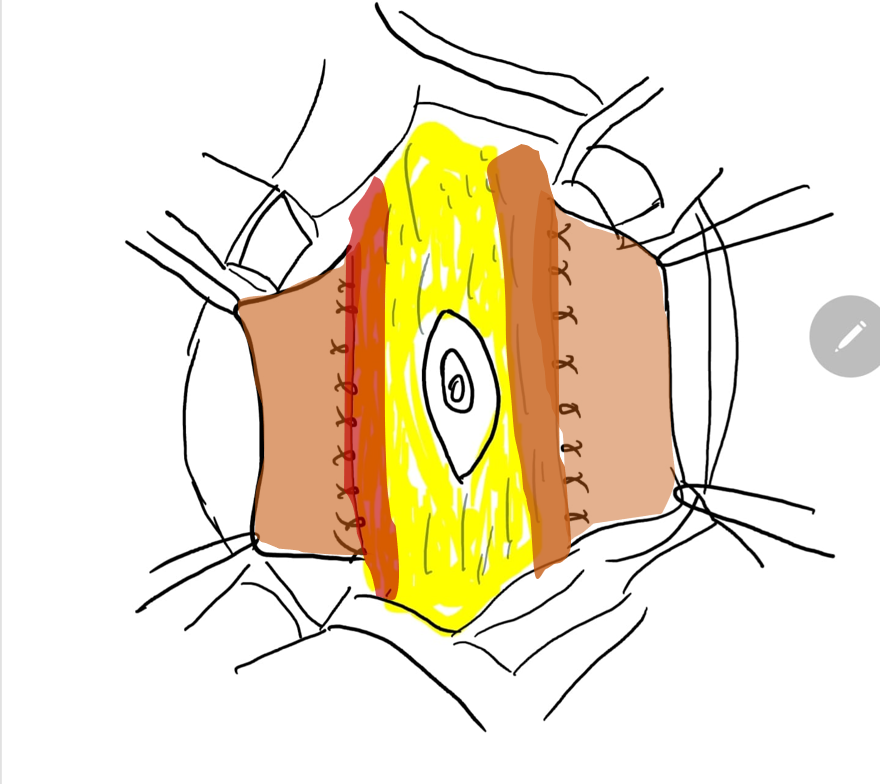
图 7 将中央筋膜条带的外侧缘缝合在肌肉的深面。
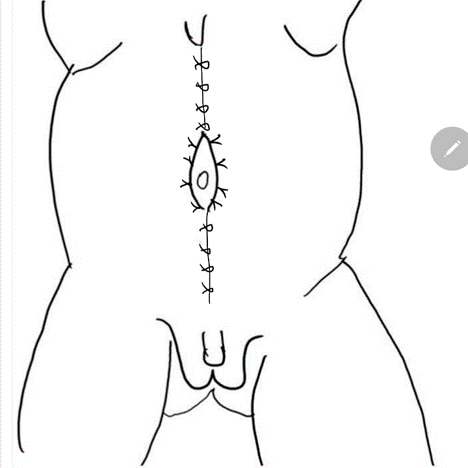
图 8 随后将外侧筋膜在脐上与脐下的正中线处固定。裤盖背心式关闭可提供额外的支持,随后将两侧的皮瓣予以关闭予以关闭。
隐睾症
双侧腹内睾丸是PBS的另一标志性特征。根据睾丸血管的长度,传统的一期睾丸固定术以及 Fowler- Stephens 睾丸固定术(可一次完成或分期进行)都是处理睾丸的潜在选择;并可根据术者偏好采用开放或腹腔镜入路实施。65,66,67 松弛的腹壁在建立和维持气腹时可能导致潜在的技术问题;在腹腔镜睾丸固定术中使用径向扩张套管针并提高气体流量可将其最小化,如 Philip 等人 所述。67,68 但需要强调的是,在不存在任何需要优先处理的心肺问题的情况下,应尽可能早期进行睾丸固定术,正如当前的 [AUA 指南](https://www.auanet.org/guidelines/guidelines/cryptorchidism-guideline) 所建议的,不应拖延至儿童期较大年龄;这样可提高生育力的机会,并提高一期手术的可能性,因为在早期进行睾丸固定术时较少需要离断睾丸血管。
综合重建
梅干腹综合征患者的综合重建包括减容膀胱成形术、远端冗余输尿管切除以及双侧输尿管锥形化再植术。69 重建可与腹壁成形术和双侧睾丸固定术同时进行,通常在2岁之前完成。这一积极的治疗策略已在许多病例系列中采用,并在肾功能稳定或改善方面显示出良好结局。
按照 Woodward 描述的入路,切开在下腹部做一宽大的切口,自第12肋骨尖端起,沿髂前上棘至耻骨,然后向上弯曲至对侧第12肋骨。这样往往可以保留拟用于重建的腹壁残余部分的节段性神经和血供。此外,在切除冗余的腹壁时,保留肌纤维最为丰富的部分。该切口对泌尿道的暴露极佳。然而,鉴于对可能中断腹壁动静脉以及侧方腹壁组织冗余的担忧,该技术的使用已减少。相反,多数外科医生更倾向采用 Monfort 入路 (图 4), (图 5), (图 6), (图 7), 和 (图 8)
沿输尿管的走行切开后腹膜。将精索血管向远端游离;如需行 Fowler-Stephens 睾丸固定术,注意不要破坏输精管的腹膜附着。沿正中用电凝切开膀胱。辨认位于外侧的输尿管开口,并分别游离每条输尿管。游离后的输尿管具有充足的长度,可切除其远端三分之一,通常也是病变最明显的部分。行规范的缩管成形术或层叠缝合(imbrication)。随后行输尿管再植,可采用经三角区(transtrigonal)或 Leadbetter Politano 技术。对已手术的输尿管置入支架,并将支架经膀胱并通过各自的腹部切口引出至体外。
在进行减容膀胱成形术之前,将睾丸连同一大片后腹膜组织一起游离。在某些情况下,睾丸可能与增大的膀胱黏连,必须细致游离以保护血供。如果在2岁之前进行重建,80%的情况下可以在保留精索血管完整的前提下行标准睾丸固定术。否则,需要进行延期或正式的Fowler-Stephens睾丸固定术
通过切除脐尿管残余和巨大的膀胱穹隆来缩小膀胱体积。可从其中一侧壁切除一条膀胱上皮条带,并采用背心覆裤式技术予以关闭。膀胱置入Malecot导管引流。先前已游离的睾丸经耻骨结节处的远端腹壁牵出。在阴囊内创建一个肉膜囊袋,并以不可吸收缝线将每个睾丸固定于囊袋内。
随后将多余的腹壁牵拉至紧张,在中线以及两侧肋椎角处做标记,以勾画拟切除的组织范围。避免过量切除腹壁至关重要。第二条切口线与先前切口平行。切除一大片全层腹壁的楔形组织。通过在髂前上棘和每个耻骨结节处放置0号或1–0号不可吸收缝线的关键缝合点开始关闭腹壁。这些缝线应包含骨的骨膜,并穿过近端相对组织边缘的全层。所产生的张力有时可允许进一步切除侧方额外的冗余腹壁。关键缝线之间的各段采用间断缝合,使腹壁组织全层对合。关闭皮下组织以防止死腔形成,并用皮下(皮内)可吸收缝线对合皮肤。
长期预后
生活质量
尽管PBS患者在生存率和生活质量方面的总体预后已有显著改善,但PBS仍深刻影响受累儿童及其照护者的健康相关生活质量。70 患儿在身体、情绪、社会及学校功能方面受损,而照护者则因长期照料慢性病儿童所承受的负担而报告总体生活质量降低。另一项研究表明,存活的PBS个体在骨科、胃肠道和心肺疾病方面的发生率显著升高,进而对其生活质量产生负面影响;而对这些非泌尿生殖系统疾病的治疗可改善其生活质量。70,71 这些研究强调,由于小儿泌尿外科医师对这类医学情况复杂患者的整体健康负有首要责任,他们不仅必须认识并准备处理PBS的远期泌尿生殖并发症,还应评估和诊断那些直接影响其内科与外科治疗及生活质量的非泌尿生殖系统合并症,并在患者经历青春期进入成年期的过程中持续加以关注。
肾功能衰竭与肾移植
多达30%的患者(通常为起初存在肾功能受损者)会在儿童期或青少年期发展为慢性肾衰竭。早发的终末期肾病(ESRD)被认为继发于肾发育不良,而较晚发生的肾衰竭则常归因于反复感染导致的肾实质损伤,以及由排空不全产生并传导至上尿路的压力增高。72 Yalcinkaya 等 报告称,患有 PBS 的男孩初次接受肾脏替代治疗的中位年龄为7岁,显著小于患有其他类型 LUTO 或肾发育不良的男性患者;其首次移植的中位年龄仅为9.3岁。73
为确保这些患者的正常生长与发育,肾移植是必要的;对于PBS患者,其移植成功率预计可与其他同龄组相当。自1976年首次报道以来,尸体供肾与有血缘关系的活体供肾均已在PBS患者中成功移植,移植时年龄范围为8个月至21岁。73,74 为避免感染并发症,移植前通常在所有病例中行双侧肾输尿管切除术。建议对下尿路进行影像学与尿动力学评估,以确保无梗阻并实现协调的排尿。为排空失代偿的膀胱而采用清洁间歇导尿并不构成肾移植的禁忌证。所有患者均应维持抗生素预防性用药。西梅腹综合征移植中的一种少见并发症是移植肾扭转。75 该扭转及其导致的移植肾丢失是由于腹壁张力不足所致。为避免这一灾难性并发症,已建议行肾固定术。在对8例接受移植的西梅腹综合征患者的回顾性研究中,与同龄对照相比,患者死亡、移植肾存活率或移植肾功能方面均无统计学差异。更近期的一项回顾性分析显示,10年移植肾存活率为47%,与同龄对照患者相比无统计学显著差异,73,74
膀胱功能
如前所述,PBS 患者的尿动力学检查通常显示顺应性正常,但感觉和收缩活动减弱。其结果是,一些患者倾向于在膀胱内潴留大量尿液,膀胱容量逐步增大,并伴有显著的排尿后残余尿。腹壁支撑不足、可能并存的多尿,以及部分患者对定时排尿的负面行为(依从性差)通常会加重这种倾向。因此,为防止膀胱进一步扩张和上尿路恶化,所有患者——即使是已接受下尿路重建和腹壁成形术者——都应尽早接受训练并持续强化,按时排空膀胱,必要时结合双次或三次排尿以及 Valsalva's 或 Credé's 动作。76 如出现进行性的排尿失衡,而上述方案不可行或无效,则必须进行间歇性导尿,可经尿道或通过 Mitrofanoff 通道。随着生长发育、本体感觉改善以及腹壁支撑增强,最终可实现有效排尿。在 Lopes et al 的系列中,接受尿路重建的患者中有 17.4% 需要经尿道或通过 Mitrofanoff 通道行间歇性导尿。少数情况下,其中一些儿童可重新获得正常排尿并良好排空膀胱。
生长与肌肉骨骼发育
对于肾功能正常的多数患者,可望获得正常的生长发育,然而,有一项系列研究发现,即便在无肾功能受损的情况下,仍有三分之一患者存在生长迟缓。34 胸廓畸形,如漏斗胸,通常在较大年龄儿童中并不影响肺功能,并且可随着生长和运动而有所改善。患有该综合征的患者可能出现影响功能和外观的肌肉骨骼异常,如脊柱侧弯、脊柱前凸、踝部和髋部异常,这些情况可能需要早期骨科干预。在一项近期研究中,年长患者及其家属被告知,通过腹壁整形术和/或骨科手术纠正肌肉骨骼问题,是医疗服务提供者改善患者生活质量的最常见方式。
性功能与生育能力
由于睾丸的激素分泌得以保留,可预期第二性征发育模式正常,但逆行射精可能损害性功能。多表现为原发性不育,其发生被认为由多种因素共同导致,包括睾丸组织学异常伴精子发生受损、输精管道的结构性缺陷,以及前列腺异常所致的逆行射精。77 然而,通过辅助生殖技术(ART)可以实现生育,尤其是在早期成功接受睾丸固定术者;已有经典型PBS成年患者通过取精技术和卵胞浆内单精子注射而获得正常活产的报道。78,79,80 亦有报道一名患有该综合征的女性发生正常妊娠,并在助产下经阴道分娩。81
结论
与大多数复杂先天畸形一样,PBS 的管理关键在于采用多学科团队协作的个体化照护方式。鉴于疾病谱广泛,梅干腹综合征的治疗需因人而异。当感染和尿路淤滞危及肾脏生长或功能时,建议采取积极的外科干预以促进引流。为改善生育力,这些婴儿的睾丸应尽早置于阴囊内,可通过腹腔镜或开放技术完成。由于下尿路尿动力学状态的变化及其对肾功能和尿路感染的影响,所有梅干腹综合征患者均需谨慎的终生泌尿学监测。
要点
- 梅干腹综合征 (PBS) 包括一组严重程度不一的异常。三大主要表现为腹壁肌缺如、双侧腹内睾丸以及泌尿道异常。
- 泌尿道的特征为不同程度的肾积水、肾发育不良、输尿管扩张迂曲、膀胱增大,以及前列腺部尿道扩张/ 巨尿道。
- 其他相关异常可累及呼吸道、胃肠道、心脏系统和肌肉骨骼系统。
- 决定长期生存最重要的因素通常是泌尿道异常的严重程度,尤其是肾发育不良的程度。
- 非梗阻性肾积水为常态。对肾功能构成最大风险的是肾脏感染而非梗阻。
- 初始泌尿外科干预旨在进行膀胱引流与充分排空,以避免尿路感染 (UTI),从而保护肾功能。
- 手术治疗包括泌尿道重建、腹壁整形术以及双侧睾丸固定术;对于年幼患儿,这些手术可分期或一期完成。
- 照护PBS患儿需要大型多学科团队协作,以帮助患儿茁壮成长、增加体重,并在需要时为泌尿外科手术做好准备。
参考文献
- Woodard S JR, E.A.. Prune belly syndrome.
- Denes FT, Lopes RI. Prune-belly syndrome. In: Partin AW, Dmochowski RR, Kavoussi LR, Peters CA, editors. Campbell wals-wein urology. 12th ed. Philadelphia: Elsevier, Inc; 2021. DOI: 10.25060/residpediatr-2018.v8n1-07.
- Grimsby GM, Harrison SM, Granberg CF, Bernstein IH, Baker LA. Impact and frequency of extra-genitourinary manifestations of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 280 281–286. DOI: 10.1016/j.jpurol.2015.06.005.
- Wong DG, Arevalo MK, Passoni NM, Iqbal NS, Jascur T, Kern AJ. Phenotypic severity scoring system and categorization for prune belly syndrome: application to a pilot cohort of 50 living patients. BJU Int 2019; 123 (1): 130–139. DOI: 10.1111/bju.14524.
- Wheatley JM, Stephens FD, Hutson JM. Prune-belly syndrome: ongoing controversies regarding pathogenesis and management. Semin Pediatr Surg 1996; 5 (2): 95–106.
- Gonzalez R, Reinberg Y, Burke B, Wells T, Vernier RL. Early bladder outlet obstruction in fetal lambs induces rental dysplasia and the prune-belly syndrome. J Pediatr Surg 1990; 25 (3): 342–345. DOI: 10.1016/0022-3468(90)90083-l.
- Ramasamy R, Haviland M, Woodard B JR, J.G.. Patterns of inheritance in familial prune belly syndrome. Urology 2005; 65 (6). DOI: 10.1016/j.urology.2004.12.050.
- Reinberg Y, Shapiro E, Manivel JC, Manley CB, Pettinato G, Gonzalez R. Prune belly syndrome in females: A triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr 1991; 118 (3). DOI: 10.1016/s0022-3476(05)82153-5.
- Balaji KC, Patil A, Townes PL, Primack W, Skare J, Hopkins T. Concordant prune belly syndrome in monozygotic twins. Urology 2000; 55 (6). DOI: 10.1016/s0090-4295(00)00452-0.
- Iqbal NS, Jascur TA, Harrison SM, Edwards AB, Smith LT, Choi ES. Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene. BMC Med Genet 2020; 21 (1). DOI: 10.1186/s12881-020-0973-x.
- Granberg CF, Harrison SM, D D. Genetic basis of prune belly syndrome: Screening for HNF1beta gene. J Urol 2012; 187 (1).
- Stephens FD, Gupta D. Pathogenesis of the prune belly syndrome. J Urol 1994; 152: 2328–2331. DOI: 10.1016/s0022-5347(17)31669-5.
- Palmer JM, Tesluk H. Ureteral pathology in the prune belly syndrome. J Urol 1974; 111 (5). DOI: 10.1016/s0022-5347(17)60050-8.
- Gearhart JP, Lee BR, Partin AW, Epstein JI, Gosling JA, Kogan BA. A quantitative histological evaluation of the dilated ureter of childhood. II: Ectopia, posterior urethral valves and the prune belly syndrome. J Urol 1995; 153 (1). DOI: 10.1097/00005392-199501000-00069.
- Snyder HM, Harrison NW, Whitfield HN, Williams I. Urodynamics in the prune belly syndrome. Br J Urol 1976; 48 (7): 663–670. DOI: 10.1111/j.1464-410x.1976.tb06716.x.
- Kinahan TJ, Churchill BM, McLorie GA, Gilmour RF, Khoury AE. The efficiency of bladder emptying in the prune belly syndrome. J Urol 1992; 148 (2 Pt 2): 600–603. DOI: 10.1016/s0022-5347(17)36665-x.
- Favorito LA, Pires RS, Gallo CM, Sampaio FJB. Study of prostate growth in prune belly syndrome and anencephalic fetuses. J Pediatr Surg 2020; 55 (10): 2221–2225. DOI: 10.1016/j.jpedsurg.2019.10.054.
- Volmar KE, Fritsch MK, Perlman EJ, Hutchins GM. Patterns of congenital lower urinary tract obstructive uropathy: Relation to abnormal prostate and bladder development and the prune belly syndrome. Pediatr Dev Pathol 2001; 4 (5). DOI: 10.1007/s10024001-0042-1.
- Deklerk DP, Scott WW. Prostatic Maldevelopment in the prune belly syndrome: A defect in prostatic stromal-epithelial interaction. J Urol 1978; 120 (3). DOI: 10.1016/s0022-5347(17)57168-2.
- Cabral B, Majidi A, Gonzalez R. Ectopic vasa deferentia in an infant with the prune belly syndrome. J Urol 1988; 94 (4).
- Kroovand RL, Al-Ansari RM, Perlmutter AD. Urethral and genital malformations in prune belly syndrome. J Urol 1982; 127 (1): 94–96. DOI: 10.1016/s0022-3468(82)80571-x.
- Berdon WE, Baker DH, Wigger HJ, Blanc WA. The radiologic and pathologic spectrum of the prune belly syndrome. The importance of urethral obstruction in prognosis. Radiol Clin North Am 1977; 15 (1).
- Hoagland MH, Hutchins GM. Obstructive lesions of the lower urinary tract in the prune belly syndrome. J Urol 1987; 138 (2). DOI: 10.1016/s0022-5347(17)43220-4.
- Volmar KE, Nguyen TC, Holcroft CJ, Blakemore KJ, Hutchins GM. Phimosis as a cause of the prune belly syndrome: Comparison to a more common pattern of proximal penile urethra obstruction. Virchows Arch 2003; 442 (2). DOI: 10.1007/s00428-002-0730-x.
- Beasley S, Bettenay F, Hutson J. The anterior urethra provides clues to the aetiology of prune belly syndrome. Pediatr Surg Int 1988; 3–3(2–3. DOI: 10.1007/bf00182775.
- Sellers BB, McNeal R, Smith RV, Griswold WR, Mendoza SA. Congenital megalourethra associated with prune belly syndrome. J Urol 1976; 116 (6). DOI: 10.1016/s0022-5347(17)59027-8.
- Uehling DT, Zadina SP, Gilbert E. Testicular histology in triad syndrome. Urology 1984; 23 (4). DOI: 10.1016/0090-4295(84)90141-9.
- Massad CA, Cohen MB, Kogan BA, Beckstead JH. Morphology and histochemistry of infant testes in the prune belly syndrome. J Urol 1991; 146 (6): 1598–1600. DOI: 10.1016/s0022-5347(17)38178-8.
- Orvis BR, Bottles K, Kogan BA. Testicular histology in fetuses with the prune belly syndrome and posterior urethral valves. J Urol 1988; 139 (2): 335–337. DOI: 10.1016/s0022-5347(17)42403-7.
- Sayre R, Stephens R, Chonko AM. Prune belly syndrome and retroperitoneal germ cell tumor. Am J Med 1986; 81 (5). DOI: 10.1016/s0022-5347(17)44221-2.
- Woodhouse CR, Ransley PG. Teratoma of the testis in the prune belly syndrome. Br J Urol 1983; 55 (5).
- Mininberg DT, Montoya F, Okada K, Galioto F, Presutti R. Subcellular muscle studies in the prune belly syndrome. J Urol 1973; 109 (3). DOI: 10.1097/00006534-197311000-00052.
- Afifi AK, Rebeiz J, Mire J, Andonian SJ, Kaloustian VM. The myopathology of the prune belly syndrome. J Neurol Sci 1972; 15 (2). DOI: 10.1016/0022-510x(72)90003-2.
- Loder RT, Guiboux JP, Bloom DA, Hensinger RN. Musculoskeletal aspects of prune-belly syndrome. Description and Pathogenesis Am J Dis Child 1992; 146 (10): 1224–1229. DOI: 10.1001/archpedi.1992.02160220110034.
- Geary DF, MacLusky IB, Churchill BM, McLorie G. A broader spectrum of abnormalities in the prune belly syndrome. J Urol 1986; 135 (2). DOI: 10.1016/s0022-5347(17)45627-8.
- Alford BA, Peoples WM, Resnick JS, L’Heureux PR. Pulmonary complications associated with the prune-belly syndrome. Radiology 1978; 129 (2). DOI: 10.1148/129.2.401.
- Wright B JR, RF N, JC P, ET S, ME S, L.E.. Gastrointestinal malformations associated with prune belly syndrome: Three cases and a review of the literature. Pediatr Pathol 1986; 5 (3–4). DOI: 10.3109/15513818609068868.
- Salihu HM, Tchuinguem G, Aliyu MH, Kouam L. Prune belly syndrome and associated malformations. A 13-year experience from a developing country. West Indian Med J 2003; 52 (4).
- Brinker MR, Palutsis RS, Sarwark JF. The orthopaedic manifestations of prune-belly (Eagle-Barrett) syndrome. J Bone Joint Surg 1995; 77 (2). DOI: 10.2106/00004623-199502000-00012.
- Cazorla E, Ruiz F, Abad A, Monleon J. Prune belly syndrome: Early antenatal diagnosis. Eur J Obstet Gynecol Reprod Biol 1997; 72 (1). DOI: 10.1016/s0301-2115(96)02664-4.
- Fitzsimons RB, Keohane C, Galvin J. Prune belly syndrome with ultrasound demonstration of reduction of megacystis in utero. Br J Radiol 1985; 58 (688). DOI: 10.1259/0007-1285-58-688-374.
- Aaronson IA. Posterior urethral valve masquerading as the prune belly syndrome. Br J Urol 1983; 55 (5). DOI: 10.1016/s0022-3468(84)80160-8.
- Tonni G, Ida V, Alessandro V, Bonasoni MP. Prune-belly syndrome: case series and review of the literature regarding early prenatal diagnosis, epidemiology, genetic factors, treatment, and prognosis. Fetal Pediatr Pathol 2013; 31 (1): 13–24. DOI: 10.3109/15513815.2012.659411.
- Yamamoto H, Nishikawa S, Hayashi T, Sagae S, Kudo R. Antenatal diagnosis of prune belly syndrome at 11 weeks of gestation. J Obstet Gynaecol Res 2001; 27 (1). DOI: 10.1111/j.1447-0756.2001.tb01213.x.
- Seidel NE, Arlen AM, Smith EA, Kirsch AJ. Clinical manifestations and management of prune-belly syndrome in a large contemporary pediatric population. Urology 2015; 85 (1). DOI: 10.1016/j.urology.2014.09.029.
- Woods AG, Brandon DH. Prune belly syndrome. A focused physical assessment. Adv Neonatal Care 2007; 7 (3): 144–145.
- Arlen AM, Nawaf C, Kirsch AJ. Prune belly syndrome: current perspectives. Pediatric Health Med Ther 2019; 10: 75–81. DOI: 10.2147/phmt.s188014.
- Rushton HG, Majd M, Chandra R, Yim D. Evaluation of 99M technetium-dimercapto-succinic acid renal scans in experimental acute pyelonephritis in piglets. J Urol 1988; 140 (5). DOI: 10.1016/s0022-5347(17)41992-6.
- Garcia-Roig ML, Grattan-Smith JD, Arlen AM, Smith EA, Kirsch AJ. Detailed evaluation of the upper urinary tract in patients with prune belly syndrome using magnetic resonance urography. J Pediatr Urol 2016; 12 (2). DOI: 10.1016/j.jpurol.2015.11.008.
- Kaplan BS. In utero intervention in prune-belly syndrome. Pediatr Nephrol 1999; 13 (2). DOI: 10.1007/s004670050581.
- Makino Y, Kobayashi H, Kyono K, Oshima K, Kawarabayashi T. Clinical results of fetal obstructive uropathy treated by vesicoamniotic shunting. Urology 2000; 55 (1). DOI: 10.1016/s0090-4295(99)00403-3.
- Burbige KA, Amodio J, Berdon WE, Hensle TW, Blanc W, Lattimer JK. Prune belly syndrome: 35 years of experience. J Urol 1987; 137 (1). DOI: 10.1016/s0022-5347(17)43880-8.
- Fallat ME, Skoog SJ, Belman AB, Eng G, Randolph JG. The prune belly syndrome: A comprehensive approach to management. J Urol 1989; 142 (3). DOI: 10.1016/s0022-5347(17)38895-x.
- Tank ES, McCoy G. Limited surgical intervention in the prune belly syndrome. J Pediatr Surg 1983; 18 (6). DOI: 10.1016/s0022-3468(83)80004-9.
- Woodhouse CRJ, Kellett MJ, Williams DI. Minimal surgical interference in the prune belly syndrome. Br J Urol 1979; 51 (6). DOI: 10.1111/j.1464-410x.1979.tb03582.x.
- Woodard P JR, T.S.. Reconstruction of the urinary tract in prune belly uropathy. J Urol 1978; 119 (6). DOI: 10.1016/s0022-5347(17)57644-2.
- Bukowski TP, Perlmutter AD. Reduction cystoplasty in the prune belly syndrome: a long-term followup. J Urol 1994; 152 (6 Pt 1): 2113–2116. DOI: 10.1016/s0022-5347(17)32333-9.
- Wille MA, Jayram G, Gundeti MS. Feasibility and early outcomes of robotic-assisted laparoscopic Mitrofanoff appendicovesicostomy in patients with prune belly syndrome. BJU Int 2011; 109 (1). DOI: 10.1111/j.1464-410x.2011.10317.x.
- Ehrlich RM, Lesavoy MA, Fine RN. Total abdominal wall reconstruction in the prune belly syndrome. J Urol 1986; 136: 282–285. DOI: 10.1016/s0022-5347(17)44842-7.
- Randolph J, Cavett C, Eng G. Abdominal wall reconstruction in the prune belly syndrome. J Pediatr Surg 1981; 16 (6): 960–964. DOI: 10.1016/s0022-5347(17)52848-7.
- Monfort G, Guys JM, Bocciardi A, Coquet M, Chevallier D. A novel technique for reconstruction of the abdominal wall in the prune belly syndrome. J Urol 1991; 146 (2 Pt 2): 639–640. DOI: 10.1016/s0022-5347(17)37880-1.
- Denes FT, Lopes RI, Oliveira LM, Tavares A, Srougi M. Modified abdominoplasty for patients with the Prune Belly syndrome. Urology 2014; 83 (2): 451–454. DOI: 10.1016/j.urology.2013.09.031.
- Fearon JA, Varkarakis G. Dynamic abdominoplasty for the treatment of prune belly syndrome. Plast Reconstr Surg 2012; 130 (3). DOI: 10.1097/prs.0b013e31825dc170.
- Franco I. Laparoscopic assisted modification of the firlit abdominal wall plication. J Urol 2005; 174 (1). DOI: 10.1097/01.ju.0000161214.65458.c2.
- Patil KK, Duffy PG, Woodhouse CR, Ransley PG. Long-term outcome of Fowler-Stephens orchiopexy in boys with prune belly syndrome. J Urol 2004; 171 (4): 1666–1669. DOI: 10.1097/01.ju.0000118139.28229.f5.
- Yu T-J, Lai M-K, Chen W-F, Wan Y-L. Two-stage orchiopexy with laparoscopic clip ligation of the spermatic vessels in prune-belly syndrome. J Pediatr Surg 1995; 30 (6). DOI: 10.1016/0022-3468(95)90768-8.
- Philip J, Mullassery D, Craigie RJ, Manikandan R, Kenny SE. Laparoscopic orchidopexy in boys with prune belly syndrome—Outcome and technical considerations. J Endourol 2011; 25 (7). DOI: 10.1089/end.2010.0257.
- Saxena AK, Brinkmann OA. Unique features of prune belly syndrome in laparoscopic surgery. J Am Coll Surg 2007; 205 (2). DOI: 10.1016/j.jamcollsurg.2007.03.008.
- Lopes RI, Tavares A, Srougi M, Dénes FT. 27 years of experience with the comprehensive surgical treatment of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 276 271–277. DOI: 10.1016/j.jpurol.2015.05.018.
- Arlen AM, Kirsch SS, Seidel NE, Garcia-Roig M, Smith EA, Kirsch AJ. Health-related quality of life in children with Prune belly Syndrome and their caregivers. Urology 2016; 87 (224). DOI: 10.1016/j.urology.2015.09.028.
- Lesavoy MA, Chang EI, Suliman A, Taylor J, Kim SE, Ehrlich RM. Long-term follow-up of total abdominal wall reconstruction for prune belly syndrome. Plast Reconstr Surg 2012; 129 (1). DOI: 10.1097/prs.0b013e3182362091.
- Reinberg Y, Manivel JC, Pettinato G, Gonzalez R. Development of renal failure in children with the prune belly syndrome. J Urol 1991; 145 (5): 1017–1019. DOI: 10.1016/0022-3468(92)90159-5.
- Alcinkaya F, Bonthuis M, Erdogan BD, Stralen KJ, Baiko S, Chehade H, et al.. Outcomes of renal replacement therapy in boys with prune belly syndrome: findings from the ESPN/ERA-EDTA Registry. Pediatr Nephrol 2018; 33 (1): 117–124. DOI: 10.1007/s00467-017-3770-9.
- Fontaine E, Salomon L, Gagnadoux MF, Niaudet P, Broyer M, Beurton D. Long-term results of renal transplantation in children with the prune-belly syndrome. J 1997; Urol.158(3 Pt 1):892-4. DOI: 10.1097/00005392-199709000-00067.
- Marvin RG, Halff GA, Elshihabi I. Renal allograft torsion associated with prune-belly syndrome. Pediatr Nephrol 1995; 9 (1). DOI: 10.1007/bf00858981.
- Smith CA, Smith EA, Parrott TS, Broecker BH, Woodard JR. Voiding function in patients with the prune-belly syndrome after Monfort abdominoplasty. J Urol 1998; 159 (5). DOI: 10.1016/s0022-3468(99)90298-1.
- Woodhouse CR, Snyder HM. Testicular and sexual function in adults with prune belly syndrome. J Urol 1985; 133 (4): 607 9. DOI: 10.1016/s0022-5347(17)49108-7.
- Halpern JA, Das A, Brannigan RE. Successful sperm retrieval in prune belly syndrome. Asian J Urol 2020; 7 (4): 376–378. DOI: 10.1016/j.ajur.2019.07.004.
- Kolettis PN, Ross JH, Kay R, Thomas AJ. Sperm retrieval and intracytoplasmic sperm injection in patients with prune-belly syndrome. Fertil Steril 1999; 72 (5). DOI: 10.1016/s0015-0282(99)00388-x.
- Fleming SD, Varughese E, Hua VK, Robertson A, Dalzell F, Boothroyd CV. Normal live births after intracytoplasmic sperm injection in a man with the rare condition of Eagle-Barrett syndrome (prune-belly syndrome. Fertil Steril 2013; 100 (6). DOI: 10.1016/j.fertnstert.2013.07.1994.
- Hillman RT, Garabedian MJ, Wallerstein RJ. Pregnancy outcome in a woman with prune belly syndrome. BMJ Case Reports 2012. DOI: 10.1136/bcr-2012-006490.
最近更新时间: 2025-09-22 08:00