
27: Prune Belly Syndrome
This chapter will take approximately 27 minutes to read.
Introduction
Prune belly syndrome (PBS) otherwise referred to as Eagle-Barrett syndrome, is a rare multisystem condition typically characterized by constellation of anomalies with variable degrees of severity, with the major findings being deficiency of the abdominal musculature, bilateral intra-abdominal testes and anomalous urinary tract.1,2Additional associated non-genitourinary anomalies involve the respiratory tract, gastrointestinal tract, cardiac system and musculoskeletal system.3 Disease severity exists along a broad continuum with some children not surviving the new-born period and others being minimally affected.4
Embryology
Several predominant theories have been proposed regarding the embryogenesis of PBS though none of them has universal acceptance, and there is some overlap between them. The four main theories are 1, an early in utero posterior urethral obstruction (probably a hypoplastic/dysplastic prostate or abnormal urethra), resulting in proximal bladder, ureteral and renal dilation with associated poor abdominal wall development 2, a primary defect in the lateral plate mesoderm, which is the precursor of the ureters, bladder, prostate, urethra, and gubernaculum 3, an intrinsic defect of the urinary tract leading to ureteral dilation and fetal ascites and 4, a yolk sac defect.5,6
Epidemiology
The incidence of PBS has been reported to be 1 in 29,000 to 1 in 40,000 live births, similar to that of bladder exstrophy with 95% of affected patients being males.1,2,7 Females represent less than 5% of all PBS cases, and they present with abdominal wall deficiency and a dilated, dysmorphic urinary tract without any associated gonadal anomaly.8 A higher incidence is noted in twins, blacks, and children born to younger mothers.9 Although PBS often presents as a sporadic event, a genetic contribution is also suspected with mutations in genes encoding smooth muscle transcription factors, contractile filaments and neural enzymes/receptors.10,11
Clinical Features and Pathogenesis
Genitourinary Anomalies
Kidneys
The spectrum of renal abnormalities in PBS patients extends from normal renal parenchyma to dysplasia. Around half the cases have renal dysplasia which is usually of the Potter type II and IV varieties and this can vary between both sides also.1,2 While the type II variety with few nephrons and parenchymal disorganization is more indicative of a renal mesenchymal defect, whereas the type IV variety with cortical and tubular cysts is associated with outlet obstruction, where there has been no urachal decompression.12 Though the renal collecting system is characteristically dilated, often to a severe degree, the degree of dilation, however, does not correlate with the degree of renal dysplasia and calyceal morphology may be well preserved, even in the presence of massive dilatation. Though, hydronephrosis is usually non obstructive, primary or secondary ureteropelvic junction obstruction can also occur. However, it is important to note that renal infection rather than obstruction that poses the greatest risk to renal function in these patients.1,2
Ureters
The ureters are typically dilated, tortuous and redundant. Though the massive dilation and stenosis can occur at all levels, the proximal (upper) portions of the ureters are usually less abnormal than the distal segments. Vesicoureteral reflux (VUR) is present in 75% of children with PBS.12 Obstruction is not common but has been reported at the ureteropelvic and ureterovesical junctions. Histologic sectioning demonstrates lack of smooth muscle cells and increase in ratio of collagen to smooth muscle in the more affected distal segments especially the refluxing ones , with more normal-appearing smooth muscle cells in the proximal segments.13 This fact is critical when ureteral reconstruction is undertaken. The decreased number of thick and thin myofibrils noted on ultrastructural examination is thought to contribute to the ineffective peristalsis because of poor ureteral wall coaptation resulting in upper tract stasis, which may lead to infection. Also, in many patients the severity of the urinary tract abnormalities is not proportional to the degree of flaccidity of the abdominal wall.14
Bladder
The bladder usually appears massively enlarged with a pseudodiverticulum at the urachus and a patent urachus in 25% to 30% of patients.12 Unlike the picture seen in obstructed bladders due to other reasons , despite being very thick, the bladder contour is smooth. On voiding, the bladder neck opens widely into a dilated prostatic urethra.1,2 Histologically, in the absence of obstruction, the bladder has an increased ratio of collagen to muscle fibers, however smooth muscle hypertrophy can be seen when there is obstruction. On cystoscopy , the trigone is splayed with the ureteral orifices displaced superiorly and laterally , possibly contributing to the high incidence of vesicoureteral reflux. Characteristic findings in a urodynamic assessment include a normal compliant bladder with delayed first sensation to void and a large capacity.15 The voiding efficiency of these bladders is variable, with some emptying to completion while others having a significant postvoid residual possibly due to a relative outlet obstruction and the detrusor muscles inability to generate sufficient pressure during voiding in these patients.16 The term “unbalanced” voiding is usually used to refer to the former situation where a relative outflow resistance prevents effective bladder emptying.1,2 Though 50% of prune-belly patients void spontaneously with normal voiding pressures, flow rates and low postvoid residuals, it has been observed that deterioration of balanced voiding can occur with growth, resulting in significant postvoid residuals, emphasizing the need for periodic assessment.16
Prostate and Accessory Sex Organs
Posterior urethral dilatation is due to prostatic hypoplasia, probably related to abnormal mesenchymal-epithelial development.12,13,14,15,16,17,18,19 Histological sections reveal few prostatic cellular elements, with a reduced epithelial and smooth muscle cells and increased connective tissue cells.19 About 20% of patients have been observed to have also obstructive lesions in the distal posterior urethra in the form of urethral atresia, valves, urethral stenosis, urethral membrane and urethral diverticulum. The lack of prostatic parenchymal tissue can lead to an angulation of the urethra during voiding, which was referred to as type IV valve by Stephens.12 Prostatic hypoplasia and the associated incompetent bladder neck is also thought to be a factor in the ejaculatory failure/ retrograde ejaculation in patients with PBS.1,2 The accessory sex organs like the vas deferens and seminal vesicles are often atretic, although either may be dilated or thickened. The epididymis may be poorly attached to the testis and there may also be lack of continuity between the efferent ductules and the rete testis.20
Anterior Urethra
Although the anterior urethra of the PBS child is usually normal, several anomalies of the urethral segment have been reported; the most common of which are urethral atresia or hypoplasia and megalourethra.21,22,23,24,25 It has been postulated that urethral atresia or hypoplasia occurs because the urethra is unused, rather than malformed. Urethral atresia is often lethal if it’s not associated with decompression in the form of a patent urachus or sometimes even spontaneous bladder rupture with fistula formation.22
An unique feature in PBS is the occurrence of megalourethra.26 Transient in utero obstruction of the junction between the glanular and penile urethra has been proposed to cause megalourethra. Two variations of megalourethra are seen in patients with PBS-namely the fusiform (Figure 1) and the scaphoid (Figure 2), (Figure 3) variety.24 The fusiform type is a deficiency of the corpus cavernosum, as well as the spongiosum probably resulting from a mesenchymal deficiency of the urethral folds and is clinically seen as dilatation of entire phallus during voiding. On the other hand, the scaphoid variety which probably arises from a mesenchymal deficiency of the urethral supportive tissues leading to deficiency of the spongiosum only with preservation of the glans and corpora cavernosa, is characterised by dilatation of ventral urethra during voiding.26
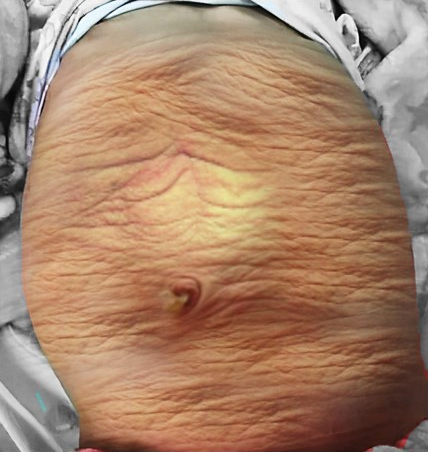
Figure 1 Clinical photograph of a child with prune belly syndrome
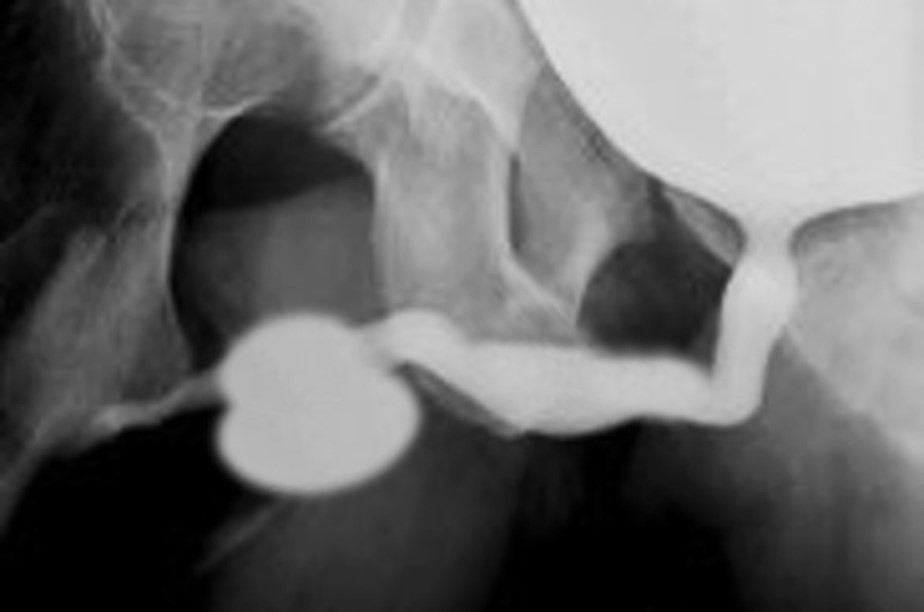
Figure 2 Fusiform megalourethra
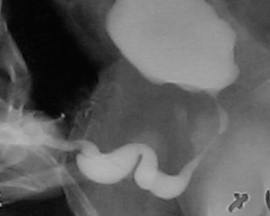
Figure 3 Scaphoid megalourethra
Testes
Bilateral intra-abdominal testes lying over the iliac vessels and adjacent to the dilated ureters are considered one of the typical findings in PBS.1,2 Although mechanical forces such as a mechanical obstruction from megacystis and poor intra-abdominal pressure due to abdominal wall abnormalities have long been hypothesized as the etiology for this failure of testicular descent, the fact that some patients with the typical urinary tract and abdominal musculature anomalies may have descended testes raises some doubt about pure mechanical factors.1,2 Regarding whether testes in children with PBS are intrinsically different from those without PBS, Pak et al compared the histology of intraabdominal testes found no difference in germ cell counts, spermatogonia and Leydig cells between PBS testes and non-PBS intraabdominal fetal testes.27,28 However, Orvis et al noted decreased numbers of spermatogonia and Leydig cell hyperplasia in fetal PBS testes implying an intrinsic testicular abnormality.29 Regarding the risk of malignancy in these testes, although the risk may be relatively low due to the lack of germinal epithelium, early orchidopexy and long-term follow-up are necessary to reduce the risk of testicular malignancy and enhance detection.30,31
Abdominal Wall Defect
The appearance of the abdominal wall is a characteristic feature in children with PBS. Most commonly the muscular deficiency is uneven and patchy, with the medial and inferior muscular segments typically most deficient and may be disproportional to the urinary tract abnormalities. The most severely affected areas may have skin, subcutaneous fat and a single fibrous layer on the peritoneum, with histological examination using electron microscopy demonstrating a nonspecific pattern of myofilament disarray, Z line disorganization and mitochondrial proliferation.32,33 In spite of these abdominal wall issues, good wound healing without a tendency toward infections or incisional hernias is noted in these children. The typical appearance at birth is that of wrinkled, redundant skin with an abdomen that bulges in the flanks. As the patients grow, some of them show improved abdominal tone and appearance with fewer wrinkles, whereas in others the abdomen takes on more of a pot-belly appearance.1,2 Gait is usually not affected, although it may be delayed, and the children tend to roll to their side and use their arms to sit from a supine position. The poor support of the lower chest wall can lead to flaring of the costal margin and the resulting compromised cough effectiveness make these children more vulnerable to respiratory illness.
Extra-Genitourinary Abnormalities
Of all children with PBS, 75% have non–urinary tract abnormalities.1,2,4 After the obvious abdominal wall defect, the most common issues are cardiac, pulmonary, and orthopedic (Table 1).34,35,36,37,38,39,40 In addition to these organ specific morbidities, nearly 50% of children with PBS are born premature, which significantly contributes to comorbidities.
Table 1 Common extra genitourinary anomalies seen in prune belly syndrome
| System | Common defects | Remarks |
|---|---|---|
| Cardiac Anomalies (10%) | Patent ductus arteriosus, atrial and ventricular septal defects, ventricular septal defect, and tetralogy of Fallot | Cardiac abnormalities at birth may take precedence over urologic issues. |
| Pulmonary Abnormalities (55%) | Pulmonary hypoplasia Pneumothorax and pneumomediastinum can be seen with or without pulmonary hypoplasia | Pulmonary hypoplasia can result from severe oligohydramnios related to renal dysplasia or severe bladder outlet obstruction and may result in new-born demise. In nearly half of new-borns with PBS, intubation and mechanical ventilatory support will be required with its attendant morbidities. The lack of ability to generate significant intra-abdominal pressure and associated musculoskeletal abnormalities such as scoliosis and rib cage abnormalities, most commonly pectus excavatum, may contribute to increased risk of pneumonia and lobar atelectasis in these children . |
| Gastrointestinal Abnormalities (30%) | Anomalies result from incomplete rotation of the midgut giving way to a wide mesentery, which results in increased bowel mobility with intestinal malrotation, volvulus, atresias, and stenosis. Splenic torsion related to abnormal mesenteric fixation has also been reported. Omphalocele, gastroschisis, situs inversus abdominis, VACTERL (vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities) and anorectal abnormalities have been reported. | With a limited ability to generate intra-abdominal pressure as a result of the abdominal muscular hypoplasia, constipation becomes a lifelong problem and may lead to acquired megacolon. |
| Orthopaedic Abnormalities (65%) | Dimpling of the lateral aspect of the knees is a common finding in oligohydramnios and is frequently seen in patients with PBS along with other defects like talipes equinovarus (26%), hip dysplasia (5%), and congenital scoliosis (4%). Lower extremity hypoplasia, absence, or amputation. | In view of the fact that most of these musculoskeletal defects are unilateral, the most likely cause of these defects is the compressive effect of oligohydramnios. Another proposed mechanism for lower limb anomalies has been compression of the external iliac vessels by the distended bladder, thereby compromising the blood supply to the lower extremities Also, some of these malformations such as renal osteodystrophy, dislocation of the hip, scoliosis and pectus excavatum or carinatum tend to manifest or worsen with growth. |
Diagnosis and Evaluation
Prenatal
Though the diagnosis of PBS on ultrasound has been reported as early as 11-12 weeks of pregnancy, the diagnostic accuracy varies from 30% to 85% as PBS presents prenatally with many sonogram findings comparable to that of lower urinary tract obstruction (LUTO) due to other causes including posterior urethral valves, ureterocele and urethral atresia.40,41,42,43,44 Prenatal diagnosis of PBS should be considered whenever the following ultrasound anomalies are clearly identified: oligohydramnios, urinary abnormalities (dilatation of the urinary tract, megacystis, bilateral hydroureteronephrosis) and the absence of abdominal musculature. Early diagnosis not only improves survival by prompting the treating clinician to conduct the delivery in a tertiary center for prompt multidisciplinary management of the affected newborns, but also gives the option of voluntary termination, if desired to the expectant parents.43
Newborn
The appearance of the abdominal wall is diagnostic in the new-born, regardless of whether the diagnosis was known prenatally. Children affected with PBS present an unique challenge and require prompt multidisciplinary care from a team, comprising a neonatologist, urologist, nephrologist as well as cardiologist and pediatric orthopedician when indicated. Given the wide range of disease severity, a multisystem evaluation to rule out significant cardiac, pulmonary, or other associated malformations must take place in the neonatal period. An immediate chest x-ray is necessary to exclude associated pneumothorax and pneumomediastinum. As is the case with LUTO, the initial postnatal course is dictated by the severity of comorbidities such as pulmonary hypoplasia with worse outcomes in those with prematurity and severe pulmonary hypoplasia.45,46,47
Antenatal oligohydramnios in these infants indicate impaired renal function. Initial creatinine measurements reflect maternal renal function and repetitive sampling is necessary. If after 48–72 hours the serum creatinine is greater than 1.0 in the term infant or 1.5 in the preterm infant, a degree of renal insufficiency is present. A progressive rise in serum creatinine over the first few weeks of life, portends a poor outcome. If initial nadir creatinine is less than 0.7 mg/dL, then subsequent renal failure is unlikely.48 Urine is sent for baseline culture and antibiotic prophylaxis is initiated. Measurement of serum and urinary electrolytes at birth can be helpful in assessing sodium conservation, implying adequate renal function.
Diagnostic Evaluation
A thorough urologic evaluation proceeds after stabilization of the patient due to other comorbidities. Physical examination and ultrasonography of the urinary tract are initially required.46 Renal sonograms provide information on cortical thickness, presence of cystic changes and renal size. Sonographic examinations before and after voiding give a clue to the presence of vesicoureteral reflux and post void residual urine.
Initially, a voiding cystourethrogram (VCUG) is performed in those infants with abnormal renal function to insure that a true anatomic, urethral obstruction is not present. This “lethal variant” and the potential confusion with posterior urethral valves “masquerading” as prune belly syndrome must be diagnosed, as treatment changes dramatically. In addition, the VCUG demonstrates vesicoureteral reflux in up to 85% of patients and assesses bladder emptying. All infants are treated with antibiotics prior to sterile catheterization to decrease the risk of infection. In general, the radiologic assessment of the urinary tract is tailored by the management philosophy of the individual urologic surgeon. Contrast studies requiring risky instrumentation are avoided in those infants with good renal function.22
After initial evaluation, a mercaptoacetyltriglycine (MAG3) renal scan can provide both functional and anatomic information. Blood flow, differential renal function and drainage in response to furosemide (Lasix) can be assessed and compared with subsequent studies. The limitations of diuresis renography in demonstrating obstruction in patients with prune belly syndrome have been noted.49 A Whitaker test is occasionally necessary as the definitive study to demonstrate significant obstruction, although this is mostly of historical significance. Dimercaptosuccinic (DMSA) acid renal scan is performed rather than an excretory urogram (IVP) to evaluate renal size, tubular mass, and differential function. Comparative studies are invaluable to assess renal growth and scarring as these patients require long-term follow up. Some groups have advocated for magnetic resonance urography (MRU) in these patients due to its high resolution and excellent functional and anatomic data that allows for detailed visualization of upper tract abnormalities which may also be useful in operative planning.50 As shown in Table 1, there are three major categories of neonatal presentation as described by Woodard.1,2
Management and Outcomes
Prenatal Management
In Utero Intervention
With improved maternal–fetal advanced imaging and diagnostic techniques, prune belly syndrome can be suspected by imaging early in pregnancy. Fetal intervention usually addresses the oligohydramnios or anhydramnios that is associated with pulmonary hypoplasia, the leading cause of perinatal mortality. Criteria for intrauterine intervention includes second or third trimester gestation, severe oligohydramnios, megacystis, advanced hydronephrosis, normal karyotype, limited extent of other detected congenital anomalies and serial favorable fetal aspirated urinalyses.51,52,53 Although several reports of vesicoamniotic shunt procedures for fetal obstructive uropathy exist, reviews have failed to document a beneficial effect of fetal intervention on subsequent renal function and also pulmonary function cannot be assured despite restoration of normal amniotic fluid levels. As the majority of patients with prune belly syndrome have no demonstrable evidence of obstruction and renal function does not correlate with the amount of dilatation present in the urinary tract, the risks and benefits of in utero intervention must be carefully weighed.53
Postnatal Management
The overall goals of management are to preserve renal function and prevent infection. The attainment of these goals are possible via a variety of therapeutic approaches, which range from watchful waiting to immediate surgical reconstruction of the urinary tract. Early surgical intervention in the neonate is avoided, unless a rising creatinine or infection occurs, requiring early vesicostomy. As shown in Table 2, management in these patients depend on the severity category at presentation.
Table 2 Presentation and management of different categories based on Woodwards criteria of neonatal presentation
| Severity Category | Presentation | Management | Follow up and Outcomes |
|---|---|---|---|
| I | Pronounced oligohydramnios secondary to renal dysplasia and/or outlet obstruction resulting in severe pulmonary hypoplasia and skeletal abnormalities | In those few infants surviving the pulmonary insult, high urinary diversion by cutaneous pyelostomy has been attempted however, recovery of renal function is not usually possible as a result of severe underlying renal dysplasia. | These neonates often expire within a few days and interventions are limited. Associated with prenatal oligohydramnios and postnatal pulmonary hypoplasia usually die in the immediate postnatal period due to pulmonary complications. |
| Potter’s facies may be present and is secondary to oligohydramnios. Features include micrognathia, wide set eyes, flattened palpebral fissures, prominent epicanthus, flattened nasal bridge, low-set ears lacking cartilage and skeletal deformities. | Only catheterization may be needed | ||
| Cases of urethral atresia are typically in this most severe category. | |||
| II | Moderate renal insufficiency and moderate-severe hydroureteronephrosis Pulmonary hypoplasia is not a prominent feature. | These children are placed on long-term prophylactic antibiotics and are serially monitored for infection and any decrease in renal function. | Lifelong postoperative surveillance needed to evaluate renal function and optimal drainage of both upper and lower tracts with functional and anatomic studies. |
| Aggressive urinary tract reconstruction is pursued with documentation of infection, failure of adequate renal growth or decreasing renal function. | |||
| However, surgical reconstruction is usually delayed until the infant is 3 months of age or more to allow pulmonary maturation. | |||
| If renal insufficiency is severe or infection is initially present, a period of urinary diversion by means of cutaneous vesicostomy is recommended. | |||
| Cutaneous ureterostomies probably add little to improve drainage. In addition, the upper ureter is anatomically and histologically the most normal and should be preserved if later tailored reconstruction is elected. | |||
| III | Mild triad features or incomplete forms | Constitute the majority of patients demonstrate good renal function, despite gross dilatation of the upper and lower urinary tract. | Similar to category 2, life-long surveillance needed. |
| Renal function is typically normal or mildly impaired and there is no pulmonary insufficiency. | Early surgical intervention to reconstruct the urinary tract is not usually necessary and these patients are managed with long term antibiotic prophylaxis and serial assessment of renal function by selective imaging techniques. | Development of urinary infections or decrease in renal function mandates further specific evaluation in the form of urodynamic assessment or diuretic renogram to document either lower tract or upper tract obstruction that may necessitate intervention. | |
| These patients benefit from early orchiopexy and abdominoplasty as correction of the abdominal wall may improve defecation and voiding efficiency. |
Operative Interventions in Prune Belly Syndrome
Urinary Tract
A characteristic finding in PBS is low-pressure dilation of the whole urinary tract, which extends from the renal pelvis proximally to the urethra distally. The bladder is typically enlarged and hypotonic, with elevated compliance and low-pressure vesicoureteral reflux (VUR) present in approximately 75% of patients. While these large volume compliant bladders allows for low pressure storage, they often demonstrate incomplete emptying secondary to reduced detrusor contractility. Therefore, initial urologic intervention is directed at bladder drainage in order to avoid urinary tract infection (UTI) and thereby preserve renal function. As such, circumcision, prophylactic antibiotics timed/double voiding are often implemented to decrease the incidence of UTI and protect the upper tracts from further insult.54,55,56,57 When such conservative management fails to prevent infection and achieve adequate emptying, these children may require reduction cystoplasty and /or appendicovesicostomy to facilitate clean intermittent catheterization and/or antireflux surgery for managing the associated VUR.58,59,60
Abdominal Wall
Abdominal wall reconstruction where the more normal peripheral abdominal wall is advanced to support this abnormal, deficient central portion is an important consideration in the surgical management of PBS patients. In addition to cosmesis, abdominoplasty has also been shown to improve functional bladder dynamics independent of genitourinary reconstruction. Urodynamic testing is often performed preoperatively to determine the need for any concomitant bladder procedures such as appendicovesicostomy in cases of incomplete emptying and/or ureteral reimplantation when VUR is present. Traditionally, three surgical procedures with their modifications have been reported in the literature for correction of the abdominal wall defects associated with PBS.61,62,63 While the Randolph procedure involves excision of a portion of the lower abdominal wall to correct vertical fascial redundancy, the Monfort (Figure 4), (Figure 5), (Figure 6), (Figure 7), (Figure 8) and Ehrlich procedures both use vertical overlapping of the fascia to correct lateral redundancy along with strengthening of the abdominal wall. A laparoscopic approach to abdominoplasty has also been reported, proving beneficial in both assisting with the reconstruction itself as well as delineating the varying degrees of muscular deficiency.64 It is also noted that PBS patients may each display unique features with regard to the variation of severity and pattern of abdominal musculature deficiency, such as a particular area with a complete paucity of muscle. Smith and colleagues recently reported their adaptations to the Monfort procedure which allows for more individualized correction of abdominal wall laxity and positioning the umbilicus to a more anatomically correct location.
Figure 4 A midline incision from xiphoid to pubis circumscribing umbilicus.
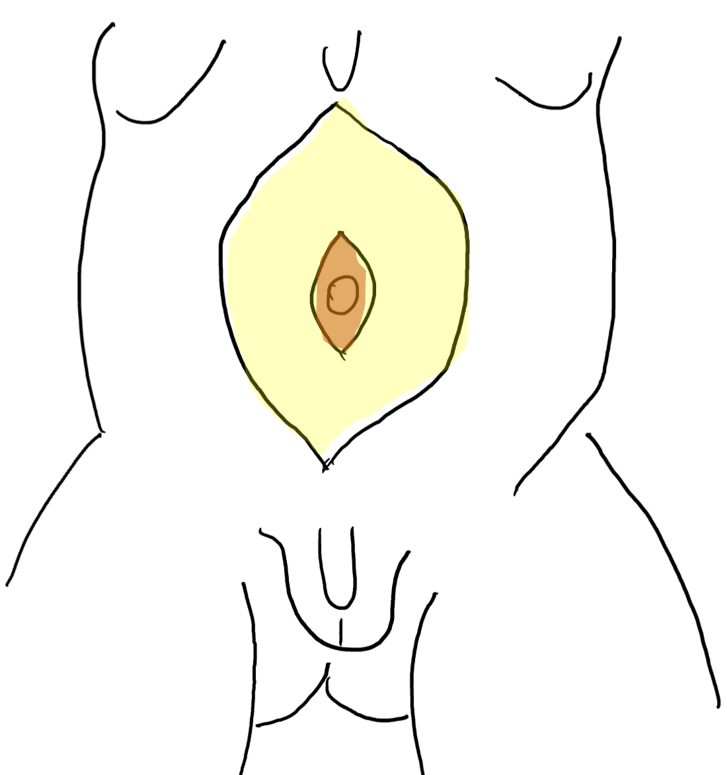
Figure 5 Raising the skin flap between subcutaneous fat and fascial layer. The umbilicus is supported by the central fascial strip.
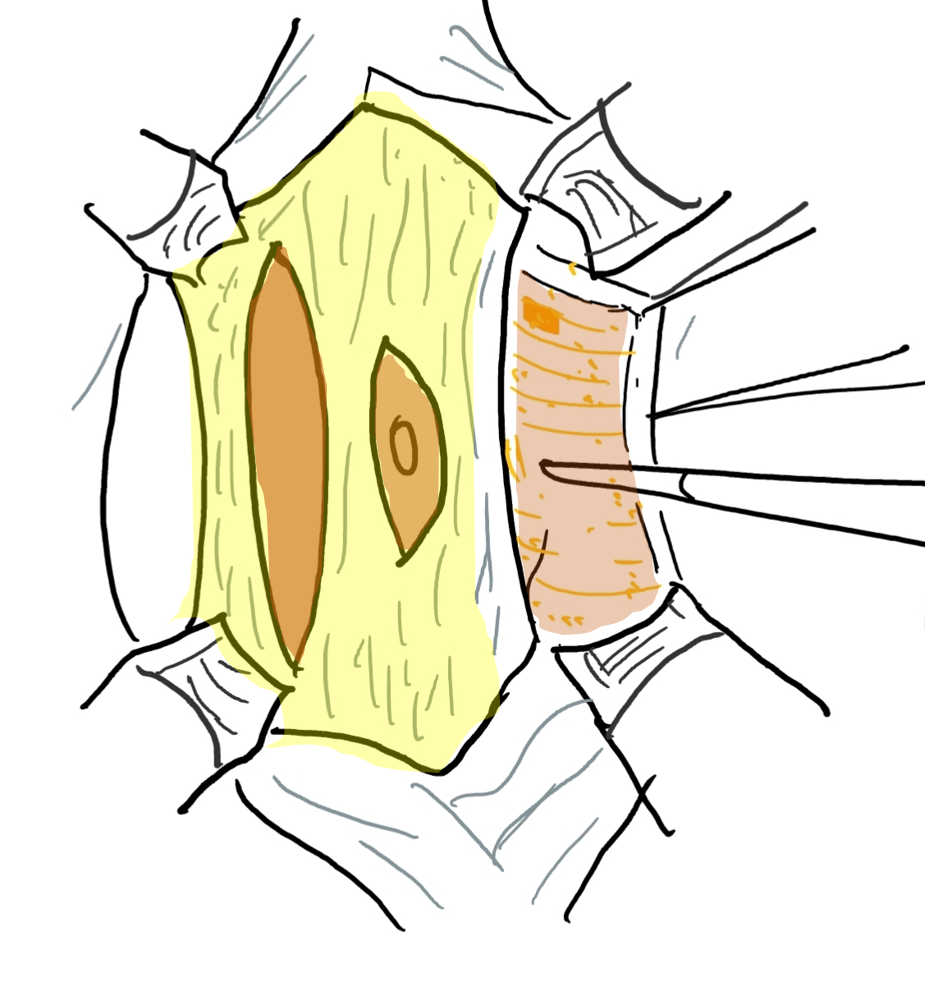
Figure 6 The fascial layer is opened through an incision lateral and parallel to the course of superior and inferior epigastric artery. The lateral extension is anterior axillary line.
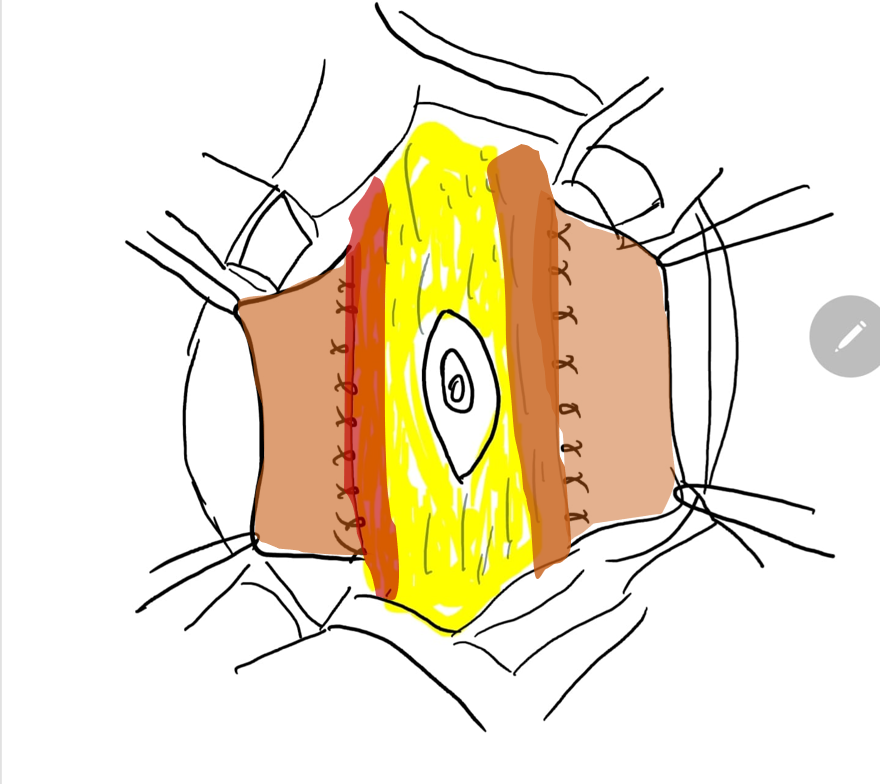
Figure 7 The lateral margin of the central fascial strip is stitched deep to the muscle.
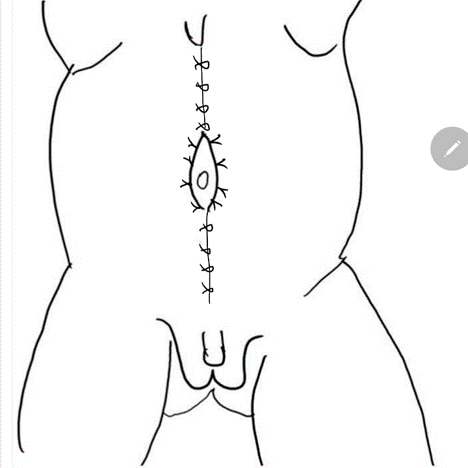
Figure 8 The lateral fascia is then secured in midline above and below the umbilicus. Pants -over-vest closure provides additional support and skin flaps from both the sides are closed are closed subsequently.
Cryptorchidism
Bilateral intra-abdominal testicles are another trademark feature of PBS. Traditional single-stage orchidopexy and single versus staged Fowler- Stephens orchidopexy are all potential options for managing the testicles, depending on gonadal vessel length, and can be performed via an open or laparoscopic approach depending upon surgeon preference.65,66,67 The lax abdominal wall can lead to potential technical issues in creating and maintaining pneumoperitoneum which can be minimized by using radially expanding trocars and high gas flow rates during laparoscopic orchidopexy as described by Philip et al.67,68 It should be emphasized, however, that in the absence of any overriding cardiopulmonary issue, orchidopexy should be performed as early as possible as recommended by the current [AUA guideline](https://www.auanet.org/guidelines/guidelines/cryptorchidism-guideline) and not delayed until older in childhood, so as to enhance the chances of fertility and improve the chances of a single stage procedure, as gonadal vessel division is less likely when orchidopexy is performed early.
Comprehensive Reconstruction
Comprehensive reconstruction in the prune belly patient involves reduction cystoplasty, resection of the redundant distal ureter and bilateral ureteral tapered reimplantation.69 Reconstruction may be combined with the performance of the abdominoplasty and bilateral orchiopexy, usually prior to two years of age. This aggressive approach has been used in many series and have shown good outcomes with respect to stable or improved renal function.
In the approach described by Woodward, a wide lower abdominal incision is made that extends from the tip of the 12th rib, along the anterior superior iliac spine to the pubis and then curves upward to the opposite 12th rib. This tends to spare the segmental nerve and vascular supply of the abdominal wall left for reconstruction. In addition, at the time of excision of the redundant abdominal wall, that portion with the most muscle fibers is preserved. The exposure of the urinary tract provided with this incision is exceptional. However, use of this technique has decreased given concerns regarding interruption of the epigastric vessels and redundant lateral abdominal tissue. Instead, most surgeons favor the Monfort approach (Figure 4), (Figure 5), (Figure 6), (Figure 7), and (Figure 8)
The posterior peritoneum is incised along the length of the ureters. The spermatic vessels are mobilized distally; care being taken not to compromise the peritoneal attachments of the vas deferens if a Fowler-Stephens orchiopexy is required. The bladder is opened in the midline with cautery. The laterally placed ureteral orifices are recognized and each ureter is mobilized. The extreme length of the mobilized ureters allows for resection of its distal third, usually the most abnormal portion. Formal tapering or imbrication is performed. The ureters are then reimplanted, using a transtrigonal or Leadbetter Politano technique. Stenting of the operated ureters is performed and these are brought out through the bladder and separate abdominal incisions.
Prior to reduction cystoplasty, the testes are mobilized with a wide apron of posterior peritoneum. In some cases, the testes may be adherent to the enlarged bladder and must be meticulously mobilized to preserve blood supply. If the reconstruction is performed prior to 2 years of age, a standard orchiopexy with intact spermatic vessels can be performed 80% of the time. Otherwise, a delayed or formal Fowler-Stephens orchiopexy will be necessary
The bladder is reduced in size by resection of the urachal remnant and the huge bladder dome. A strip of bladder epithelium can be removed from one of the lateral walls and closed by the vest-over-pants technique. The bladder is drained with a Malecot catheter. The previously mobilized testes are brought up through the distal abdominal wall at the pubic tubercle. A dartos pouch is created in the scrotum and each testis is secured in the pouch with permanent sutures.
The redundant abdominal wall is then held taut and marked in the midline and laterally at the costovertebral angles to outline the tissue to be excised. Avoidance of removal of excessive amounts of abdominal wall is critical. The second line of incision parallels the previous incision. A large wedge of full thickness abdominal wall is removed. The closure of the abdominal wall is commenced by placement of key sutures of 0 or 1–0 non-absorbable sutures at the anterior iliac spines and at each pubic tubercle. These sutures should include the periosteum of the bone and a full thickness of the proximal opposing tissue edge. The tension created occasionally allows for removal of additional lateral redundant abdominal wall. Each section between the key sutures is closed with interrupted sutures opposing the full thickness of the abdominal tissue. The subcutaneous tissue is closed to prevent dead space and the skin is approximated with a subcuticular absorbable suture.
Long Term Outcomes
Quality of Life
Though the overall outlook for the patient with PBS for survival and for quality of life has improved considerably, PBS profoundly affects health-related quality of life in affected children as well as in their caregivers.70 The patients have compromised physical, emotional, social, and school functioning and the caregivers report an overall lower quality of life because of the burdens that families with chronically ill children often face. Another study demonstrated that surviving individuals with PBS had a significantly high incidence of orthopaedic, gastrointestinal, and cardiopulmonary conditions that negatively affected their quality of life, whereas treatment of these non-GU conditions improves their lives.70,71 These studies stress the fact that, as primarily responsible for the overall health of these medically complex patients, the paediatric urologist must be aware and prepared not only to treat the late genitourinary complications of PBS but also to evaluate and diagnose the non-genitourinary comorbidities that directly affect their medical and surgical treatment and quality of life, as they pass through puberty into adulthood.
Renal Failure and Transplantation
Up to 30% of patients, generally those with initial impaired renal function, develop chronic renal failure during childhood or adolescence. Early end stage renal disease (ESRD) is thought to be secondary to renal dysplasia, whereas kidney failure occurring later is often attributed to parenchymal damage from repeated infections and the increased pressure transmitted to the upper tracts generated from incomplete emptying.72 Yalcinkaya et al reported that the median age of initial renal replacement therapy for boys with PBS was 7 years, which was significantly younger than male patients with other types of LUTO or renal dysplasia, with a median age at first transplant at just 9.3 years.73
Renal transplantation is necessary for these patients to ensure normal growth and development, and success with transplantation in patients with PBS can be expected to equal that in other age-matched groups. Since the first report in 1976, both cadaver and living-related donor kidneys have been successfully transplanted in patients with PBS, with age at transplantation ranging from 8 months to 21 years of age.73,74 Prior to transplantation, bilateral nephroureterectomies are usually performed in all cases to avoid infectious complications. Radiographic and urodynamic assessment of the lower urinary tract is recommended to insure absence of obstruction and balanced voiding. The use of clean intermittent catheterization to empty the decompensated bladder is not a contraindication to renal transplantation. All patients should be kept on antibiotic prophylaxis. An unusual complication of transplantation in prune belly syndrome is allograft torsion.75 The torsion, with resultant graft loss, was a result of lack of abdominal wall tone. Nephropexy has been recommended to avoid this disastrous complication. In a retrospective review of eight patients with prune belly syndrome who underwent transplantation, there was no statistically significant difference in patient deaths, graft survival or graft function when compared to age-matched controls. A more recent retrospective analysis of graft survival at 10 years of 47% was not statistically significantly different from age-matched controlled patients,73,74
Bladder Function
As mentioned before, urodynamic studies of patients with PBS generally show normal compliance with decreased sensibility and contractile activity. As a consequence, some patients tend to retain large volumes in the bladder, which develops progressive capacity associated to significant postvoid residual. The lack of abdominal support, the eventual association of polyuria, and the negative behaviour of some patients in regard to timed voiding usually increase this tendency. Therefore, to prevent further bladder dilation and upper urinary tract deterioration, all patients, even those who underwent lower urinary tract reconstruction and abdominoplasty, have to be trained as early as possible and constantly reinforced to empty their bladders on schedule, eventually associating it to double or triple voiding and Valsalva's or Credé's maneuvers.76 In the case of progressive unbalanced voiding, if such a program is not feasible or ineffective, intermittent bladder catheterization is mandatory, either per urethra or through a Mitrofanoff channel. Eventually, effective voiding is achieved with growth and improved proprioceptive sensation, as well as increased abdominal support. In the series of Lopes et al, 17.4% of the patients submitted to reconstruction of the urinary tract required intermittent catheterization through urethra or a Mitrofanoff channel. Rarely, some of these children reacquire normal voiding with good bladder emptying.
Growth and Musculoskeletal Development
Normal growth can be expected in most of the patients with normal renal function, although growth retardation in the absence of renal compromise was observed in one-third of the patients in one series.34 Thoracic deformities, such as pectus excavatum, usually do not compromise pulmonary function in the older child and may show some improvement with growth and exercise. Patients with the syndrome may present musculoskeletal abnormalities that impair function and cosmesis like scoliosis, lordosis, ankle and hip abnormalities that may need early orthopaedic intervention. In a recent study, older patients and their families were informed that the correction of musculoskeletal problems through abdominoplasty and/or orthopaedic surgeries was the most common way for health providers to improve their patients' quality of life.
Sexual Function and Fertility
A normal pattern of secondary sexual development can be expected as hormone production by the testes is preserved, but sexual function may be impaired by retrograde ejaculation. Primary infertility is usually the norm and if affected then this is thought to be due to a combination of testicular histologic abnormalities with impaired spermatogenesis , structural defects of the ducts, and prostatic abnormalities leading to retrograde ejaculation.77 However, paternity is achievable with assisted reproductive techniques (ART) especially those who had successful early orchidopexy and there are reports of normal live births in adult patients with classic PBS, made possible by sperm retrieval techniques and intracytoplasmic sperm injection.78,79,80 Normal pregnancy with assisted vaginal delivery has also been described in a female patient with the syndrome.81
Conclusions
As with most complex congenital anomalies, the key to management of PBS is a multidisciplinary team-based approach providing individualized care. Given the wide spectrum of disease, the treatment of prune belly syndrome is patient specific. When infection and stasis compromise renal growth or function, aggressive surgical intervention to promote drainage is advised. The testes in these infants are placed in the scrotum early for improved fertility purposes and can be accomplished by laparoscopic or open techniques. Because of the changing urodynamic condition of the lower urinary tract and its effects on renal function and urinary infection, all patients with prune belly syndrome require careful lifelong urologic surveillance.
Key Points
- Prune-belly syndrome (PBS) includes a constellation of anomalies with variable degrees of severity. The three major findings are deficiency of the abdominal musculature, bilateral intra-abdominal testes and anomalous urinary tract.
- The urinary tract is characterized by variable degrees of hydronephrosis, renal dysplasia, dilated tortuous ureters, enlarged bladder, and dilated prostatic urethra/ megalourethra.
- Additional associated anomalies involve the respiratory tract, gastrointestinal tract, cardiac system and musculoskeletal system.
- The most important determinant of long-term survival is usually the severity of the urinary tract anomaly, in particular, the degree of renal dysplasia.
- Non-obstructive hydronephrosis is the rule. It is renal infection rather than obstruction that poses the greatest risk to renal function.
- Initial urologic intervention is directed at bladder drainage and proper bladder emptying in order to avoid urinary tract infection (UTI) and thereby preserve renal function.
- Surgical management is in the form of urinary tract reconstruction, abdominoplasty and bilateral orchidopexy which can be done separately or in a single stage in younger children.
- Caring for patients with PBS requires a large multidisciplinary team approach to help these children thrive, gain weight, and be prepared for urological surgery if needed.
References
- Woodard S JR, E.A.. Prune belly syndrome.
- Denes FT, Lopes RI. Prune-belly syndrome. In: Partin AW, Dmochowski RR, Kavoussi LR, Peters CA, editors. Campbell wals-wein urology. 12th ed. Philadelphia: Elsevier, Inc; 2021. DOI: 10.25060/residpediatr-2018.v8n1-07.
- Grimsby GM, Harrison SM, Granberg CF, Bernstein IH, Baker LA. Impact and frequency of extra-genitourinary manifestations of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 280 281–286. DOI: 10.1016/j.jpurol.2015.06.005.
- Wong DG, Arevalo MK, Passoni NM, Iqbal NS, Jascur T, Kern AJ. Phenotypic severity scoring system and categorization for prune belly syndrome: application to a pilot cohort of 50 living patients. BJU Int 2019; 123 (1): 130–139. DOI: 10.1111/bju.14524.
- Wheatley JM, Stephens FD, Hutson JM. Prune-belly syndrome: ongoing controversies regarding pathogenesis and management. Semin Pediatr Surg 1996; 5 (2): 95–106.
- Gonzalez R, Reinberg Y, Burke B, Wells T, Vernier RL. Early bladder outlet obstruction in fetal lambs induces rental dysplasia and the prune-belly syndrome. J Pediatr Surg 1990; 25 (3): 342–345. DOI: 10.1016/0022-3468(90)90083-l.
- Ramasamy R, Haviland M, Woodard B JR, J.G.. Patterns of inheritance in familial prune belly syndrome. Urology 2005; 65 (6). DOI: 10.1016/j.urology.2004.12.050.
- Reinberg Y, Shapiro E, Manivel JC, Manley CB, Pettinato G, Gonzalez R. Prune belly syndrome in females: A triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr 1991; 118 (3). DOI: 10.1016/s0022-3476(05)82153-5.
- Balaji KC, Patil A, Townes PL, Primack W, Skare J, Hopkins T. Concordant prune belly syndrome in monozygotic twins. Urology 2000; 55 (6). DOI: 10.1016/s0090-4295(00)00452-0.
- Iqbal NS, Jascur TA, Harrison SM, Edwards AB, Smith LT, Choi ES. Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene. BMC Med Genet 2020; 21 (1). DOI: 10.1186/s12881-020-0973-x.
- Granberg CF, Harrison SM, D D. Genetic basis of prune belly syndrome: Screening for HNF1beta gene. J Urol 2012; 187 (1).
- Stephens FD, Gupta D. Pathogenesis of the prune belly syndrome. J Urol 1994; 152: 2328–2331. DOI: 10.1016/s0022-5347(17)31669-5.
- Palmer JM, Tesluk H. Ureteral pathology in the prune belly syndrome. J Urol 1974; 111 (5). DOI: 10.1016/s0022-5347(17)60050-8.
- Gearhart JP, Lee BR, Partin AW, Epstein JI, Gosling JA, Kogan BA. A quantitative histological evaluation of the dilated ureter of childhood. II: Ectopia, posterior urethral valves and the prune belly syndrome. J Urol 1995; 153 (1). DOI: 10.1097/00005392-199501000-00069.
- Snyder HM, Harrison NW, Whitfield HN, Williams I. Urodynamics in the prune belly syndrome. Br J Urol 1976; 48 (7): 663–670. DOI: 10.1111/j.1464-410x.1976.tb06716.x.
- Kinahan TJ, Churchill BM, McLorie GA, Gilmour RF, Khoury AE. The efficiency of bladder emptying in the prune belly syndrome. J Urol 1992; 148 (2 Pt 2): 600–603. DOI: 10.1016/s0022-5347(17)36665-x.
- Favorito LA, Pires RS, Gallo CM, Sampaio FJB. Study of prostate growth in prune belly syndrome and anencephalic fetuses. J Pediatr Surg 2020; 55 (10): 2221–2225. DOI: 10.1016/j.jpedsurg.2019.10.054.
- Volmar KE, Fritsch MK, Perlman EJ, Hutchins GM. Patterns of congenital lower urinary tract obstructive uropathy: Relation to abnormal prostate and bladder development and the prune belly syndrome. Pediatr Dev Pathol 2001; 4 (5). DOI: 10.1007/s10024001-0042-1.
- Deklerk DP, Scott WW. Prostatic Maldevelopment in the prune belly syndrome: A defect in prostatic stromal-epithelial interaction. J Urol 1978; 120 (3). DOI: 10.1016/s0022-5347(17)57168-2.
- Cabral B, Majidi A, Gonzalez R. Ectopic vasa deferentia in an infant with the prune belly syndrome. J Urol 1988; 94 (4).
- Kroovand RL, Al-Ansari RM, Perlmutter AD. Urethral and genital malformations in prune belly syndrome. J Urol 1982; 127 (1): 94–96. DOI: 10.1016/s0022-3468(82)80571-x.
- Berdon WE, Baker DH, Wigger HJ, Blanc WA. The radiologic and pathologic spectrum of the prune belly syndrome. The importance of urethral obstruction in prognosis. Radiol Clin North Am 1977; 15 (1).
- Hoagland MH, Hutchins GM. Obstructive lesions of the lower urinary tract in the prune belly syndrome. J Urol 1987; 138 (2). DOI: 10.1016/s0022-5347(17)43220-4.
- Volmar KE, Nguyen TC, Holcroft CJ, Blakemore KJ, Hutchins GM. Phimosis as a cause of the prune belly syndrome: Comparison to a more common pattern of proximal penile urethra obstruction. Virchows Arch 2003; 442 (2). DOI: 10.1007/s00428-002-0730-x.
- Beasley S, Bettenay F, Hutson J. The anterior urethra provides clues to the aetiology of prune belly syndrome. Pediatr Surg Int 1988; 3–3(2–3. DOI: 10.1007/bf00182775.
- Sellers BB, McNeal R, Smith RV, Griswold WR, Mendoza SA. Congenital megalourethra associated with prune belly syndrome. J Urol 1976; 116 (6). DOI: 10.1016/s0022-5347(17)59027-8.
- Uehling DT, Zadina SP, Gilbert E. Testicular histology in triad syndrome. Urology 1984; 23 (4). DOI: 10.1016/0090-4295(84)90141-9.
- Massad CA, Cohen MB, Kogan BA, Beckstead JH. Morphology and histochemistry of infant testes in the prune belly syndrome. J Urol 1991; 146 (6): 1598–1600. DOI: 10.1016/s0022-5347(17)38178-8.
- Orvis BR, Bottles K, Kogan BA. Testicular histology in fetuses with the prune belly syndrome and posterior urethral valves. J Urol 1988; 139 (2): 335–337. DOI: 10.1016/s0022-5347(17)42403-7.
- Sayre R, Stephens R, Chonko AM. Prune belly syndrome and retroperitoneal germ cell tumor. Am J Med 1986; 81 (5). DOI: 10.1016/s0022-5347(17)44221-2.
- Woodhouse CR, Ransley PG. Teratoma of the testis in the prune belly syndrome. Br J Urol 1983; 55 (5).
- Mininberg DT, Montoya F, Okada K, Galioto F, Presutti R. Subcellular muscle studies in the prune belly syndrome. J Urol 1973; 109 (3). DOI: 10.1097/00006534-197311000-00052.
- Afifi AK, Rebeiz J, Mire J, Andonian SJ, Kaloustian VM. The myopathology of the prune belly syndrome. J Neurol Sci 1972; 15 (2). DOI: 10.1016/0022-510x(72)90003-2.
- Loder RT, Guiboux JP, Bloom DA, Hensinger RN. Musculoskeletal aspects of prune-belly syndrome. Description and Pathogenesis Am J Dis Child 1992; 146 (10): 1224–1229. DOI: 10.1001/archpedi.1992.02160220110034.
- Geary DF, MacLusky IB, Churchill BM, McLorie G. A broader spectrum of abnormalities in the prune belly syndrome. J Urol 1986; 135 (2). DOI: 10.1016/s0022-5347(17)45627-8.
- Alford BA, Peoples WM, Resnick JS, L’Heureux PR. Pulmonary complications associated with the prune-belly syndrome. Radiology 1978; 129 (2). DOI: 10.1148/129.2.401.
- Wright B JR, RF N, JC P, ET S, ME S, L.E.. Gastrointestinal malformations associated with prune belly syndrome: Three cases and a review of the literature. Pediatr Pathol 1986; 5 (3–4). DOI: 10.3109/15513818609068868.
- Salihu HM, Tchuinguem G, Aliyu MH, Kouam L. Prune belly syndrome and associated malformations. A 13-year experience from a developing country. West Indian Med J 2003; 52 (4).
- Brinker MR, Palutsis RS, Sarwark JF. The orthopaedic manifestations of prune-belly (Eagle-Barrett) syndrome. J Bone Joint Surg 1995; 77 (2). DOI: 10.2106/00004623-199502000-00012.
- Cazorla E, Ruiz F, Abad A, Monleon J. Prune belly syndrome: Early antenatal diagnosis. Eur J Obstet Gynecol Reprod Biol 1997; 72 (1). DOI: 10.1016/s0301-2115(96)02664-4.
- Fitzsimons RB, Keohane C, Galvin J. Prune belly syndrome with ultrasound demonstration of reduction of megacystis in utero. Br J Radiol 1985; 58 (688). DOI: 10.1259/0007-1285-58-688-374.
- Aaronson IA. Posterior urethral valve masquerading as the prune belly syndrome. Br J Urol 1983; 55 (5). DOI: 10.1016/s0022-3468(84)80160-8.
- Tonni G, Ida V, Alessandro V, Bonasoni MP. Prune-belly syndrome: case series and review of the literature regarding early prenatal diagnosis, epidemiology, genetic factors, treatment, and prognosis. Fetal Pediatr Pathol 2013; 31 (1): 13–24. DOI: 10.3109/15513815.2012.659411.
- Yamamoto H, Nishikawa S, Hayashi T, Sagae S, Kudo R. Antenatal diagnosis of prune belly syndrome at 11 weeks of gestation. J Obstet Gynaecol Res 2001; 27 (1). DOI: 10.1111/j.1447-0756.2001.tb01213.x.
- Seidel NE, Arlen AM, Smith EA, Kirsch AJ. Clinical manifestations and management of prune-belly syndrome in a large contemporary pediatric population. Urology 2015; 85 (1). DOI: 10.1016/j.urology.2014.09.029.
- Woods AG, Brandon DH. Prune belly syndrome. A focused physical assessment. Adv Neonatal Care 2007; 7 (3): 144–145.
- Arlen AM, Nawaf C, Kirsch AJ. Prune belly syndrome: current perspectives. Pediatric Health Med Ther 2019; 10: 75–81. DOI: 10.2147/phmt.s188014.
- Rushton HG, Majd M, Chandra R, Yim D. Evaluation of 99M technetium-dimercapto-succinic acid renal scans in experimental acute pyelonephritis in piglets. J Urol 1988; 140 (5). DOI: 10.1016/s0022-5347(17)41992-6.
- Garcia-Roig ML, Grattan-Smith JD, Arlen AM, Smith EA, Kirsch AJ. Detailed evaluation of the upper urinary tract in patients with prune belly syndrome using magnetic resonance urography. J Pediatr Urol 2016; 12 (2). DOI: 10.1016/j.jpurol.2015.11.008.
- Kaplan BS. In utero intervention in prune-belly syndrome. Pediatr Nephrol 1999; 13 (2). DOI: 10.1007/s004670050581.
- Makino Y, Kobayashi H, Kyono K, Oshima K, Kawarabayashi T. Clinical results of fetal obstructive uropathy treated by vesicoamniotic shunting. Urology 2000; 55 (1). DOI: 10.1016/s0090-4295(99)00403-3.
- Burbige KA, Amodio J, Berdon WE, Hensle TW, Blanc W, Lattimer JK. Prune belly syndrome: 35 years of experience. J Urol 1987; 137 (1). DOI: 10.1016/s0022-5347(17)43880-8.
- Fallat ME, Skoog SJ, Belman AB, Eng G, Randolph JG. The prune belly syndrome: A comprehensive approach to management. J Urol 1989; 142 (3). DOI: 10.1016/s0022-5347(17)38895-x.
- Tank ES, McCoy G. Limited surgical intervention in the prune belly syndrome. J Pediatr Surg 1983; 18 (6). DOI: 10.1016/s0022-3468(83)80004-9.
- Woodhouse CRJ, Kellett MJ, Williams DI. Minimal surgical interference in the prune belly syndrome. Br J Urol 1979; 51 (6). DOI: 10.1111/j.1464-410x.1979.tb03582.x.
- Woodard P JR, T.S.. Reconstruction of the urinary tract in prune belly uropathy. J Urol 1978; 119 (6). DOI: 10.1016/s0022-5347(17)57644-2.
- Bukowski TP, Perlmutter AD. Reduction cystoplasty in the prune belly syndrome: a long-term followup. J Urol 1994; 152 (6 Pt 1): 2113–2116. DOI: 10.1016/s0022-5347(17)32333-9.
- Wille MA, Jayram G, Gundeti MS. Feasibility and early outcomes of robotic-assisted laparoscopic Mitrofanoff appendicovesicostomy in patients with prune belly syndrome. BJU Int 2011; 109 (1). DOI: 10.1111/j.1464-410x.2011.10317.x.
- Ehrlich RM, Lesavoy MA, Fine RN. Total abdominal wall reconstruction in the prune belly syndrome. J Urol 1986; 136: 282–285. DOI: 10.1016/s0022-5347(17)44842-7.
- Randolph J, Cavett C, Eng G. Abdominal wall reconstruction in the prune belly syndrome. J Pediatr Surg 1981; 16 (6): 960–964. DOI: 10.1016/s0022-5347(17)52848-7.
- Monfort G, Guys JM, Bocciardi A, Coquet M, Chevallier D. A novel technique for reconstruction of the abdominal wall in the prune belly syndrome. J Urol 1991; 146 (2 Pt 2): 639–640. DOI: 10.1016/s0022-5347(17)37880-1.
- Denes FT, Lopes RI, Oliveira LM, Tavares A, Srougi M. Modified abdominoplasty for patients with the Prune Belly syndrome. Urology 2014; 83 (2): 451–454. DOI: 10.1016/j.urology.2013.09.031.
- Fearon JA, Varkarakis G. Dynamic abdominoplasty for the treatment of prune belly syndrome. Plast Reconstr Surg 2012; 130 (3). DOI: 10.1097/prs.0b013e31825dc170.
- Franco I. Laparoscopic assisted modification of the firlit abdominal wall plication. J Urol 2005; 174 (1). DOI: 10.1097/01.ju.0000161214.65458.c2.
- Patil KK, Duffy PG, Woodhouse CR, Ransley PG. Long-term outcome of Fowler-Stephens orchiopexy in boys with prune belly syndrome. J Urol 2004; 171 (4): 1666–1669. DOI: 10.1097/01.ju.0000118139.28229.f5.
- Yu T-J, Lai M-K, Chen W-F, Wan Y-L. Two-stage orchiopexy with laparoscopic clip ligation of the spermatic vessels in prune-belly syndrome. J Pediatr Surg 1995; 30 (6). DOI: 10.1016/0022-3468(95)90768-8.
- Philip J, Mullassery D, Craigie RJ, Manikandan R, Kenny SE. Laparoscopic orchidopexy in boys with prune belly syndrome—Outcome and technical considerations. J Endourol 2011; 25 (7). DOI: 10.1089/end.2010.0257.
- Saxena AK, Brinkmann OA. Unique features of prune belly syndrome in laparoscopic surgery. J Am Coll Surg 2007; 205 (2). DOI: 10.1016/j.jamcollsurg.2007.03.008.
- Lopes RI, Tavares A, Srougi M, Dénes FT. 27 years of experience with the comprehensive surgical treatment of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 276 271–277. DOI: 10.1016/j.jpurol.2015.05.018.
- Arlen AM, Kirsch SS, Seidel NE, Garcia-Roig M, Smith EA, Kirsch AJ. Health-related quality of life in children with Prune belly Syndrome and their caregivers. Urology 2016; 87 (224). DOI: 10.1016/j.urology.2015.09.028.
- Lesavoy MA, Chang EI, Suliman A, Taylor J, Kim SE, Ehrlich RM. Long-term follow-up of total abdominal wall reconstruction for prune belly syndrome. Plast Reconstr Surg 2012; 129 (1). DOI: 10.1097/prs.0b013e3182362091.
- Reinberg Y, Manivel JC, Pettinato G, Gonzalez R. Development of renal failure in children with the prune belly syndrome. J Urol 1991; 145 (5): 1017–1019. DOI: 10.1016/0022-3468(92)90159-5.
- Alcinkaya F, Bonthuis M, Erdogan BD, Stralen KJ, Baiko S, Chehade H, et al.. Outcomes of renal replacement therapy in boys with prune belly syndrome: findings from the ESPN/ERA-EDTA Registry. Pediatr Nephrol 2018; 33 (1): 117–124. DOI: 10.1007/s00467-017-3770-9.
- Fontaine E, Salomon L, Gagnadoux MF, Niaudet P, Broyer M, Beurton D. Long-term results of renal transplantation in children with the prune-belly syndrome. J 1997; Urol.158(3 Pt 1):892-4. DOI: 10.1097/00005392-199709000-00067.
- Marvin RG, Halff GA, Elshihabi I. Renal allograft torsion associated with prune-belly syndrome. Pediatr Nephrol 1995; 9 (1). DOI: 10.1007/bf00858981.
- Smith CA, Smith EA, Parrott TS, Broecker BH, Woodard JR. Voiding function in patients with the prune-belly syndrome after Monfort abdominoplasty. J Urol 1998; 159 (5). DOI: 10.1016/s0022-3468(99)90298-1.
- Woodhouse CR, Snyder HM. Testicular and sexual function in adults with prune belly syndrome. J Urol 1985; 133 (4): 607 9. DOI: 10.1016/s0022-5347(17)49108-7.
- Halpern JA, Das A, Brannigan RE. Successful sperm retrieval in prune belly syndrome. Asian J Urol 2020; 7 (4): 376–378. DOI: 10.1016/j.ajur.2019.07.004.
- Kolettis PN, Ross JH, Kay R, Thomas AJ. Sperm retrieval and intracytoplasmic sperm injection in patients with prune-belly syndrome. Fertil Steril 1999; 72 (5). DOI: 10.1016/s0015-0282(99)00388-x.
- Fleming SD, Varughese E, Hua VK, Robertson A, Dalzell F, Boothroyd CV. Normal live births after intracytoplasmic sperm injection in a man with the rare condition of Eagle-Barrett syndrome (prune-belly syndrome. Fertil Steril 2013; 100 (6). DOI: 10.1016/j.fertnstert.2013.07.1994.
- Hillman RT, Garabedian MJ, Wallerstein RJ. Pregnancy outcome in a woman with prune belly syndrome. BMJ Case Reports 2012. DOI: 10.1136/bcr-2012-006490.
Last updated: 2025-09-25 12:10