
25: Anomalias de duplicação uretral e siringocele
Este capítulo levará aproximadamente 14 minutos para ler.
Introdução
As anomalias uretrais são afecções congênitas que não são raras, com algumas variações raras. Este capítulo abordará a embriologia, epidemiologia, diagnóstico, avaliação, reparo, seguimento e complicações de algumas anomalias anteriores da uretra, incluindo válvula uretral anterior (AUV), siringocele, duplicação uretral e megalouretra. Alguns acreditam até que essas anomalias, exceto a duplicação, são a mesma entidade e constituem um espectro.
Embriologia
O desenvolvimento da uretra masculina tem origem na extensão da cavidade do seio urogenital até à superfície do tubérculo genital durante a 6.ª semana de gestação. Este sulco de origem endodérmica torna-se uma placa sólida de células, que eventualmente se tubulariza no sentido proximal-distal para formar a uretra fálica. O aspeto é idêntico em fetos masculinos e femininos até à 9.ª semana e, até à 14.ª semana, a uretra masculina encontra-se completa no seu desenvolvimento. A uretra foi dividida em quatro regiões distintas, cada uma com a sua classificação anatómica própria e com o tipo celular histológico predominante da mucosa uretral. A uretra prostática e a uretra membranosa são as mais proximais, derivadas do seio urogenital e compostas por epitélio de transição delicado. Seguem-se a uretra bulbar e a uretra pendular, derivadas da placa uretral na face ventral do tubérculo genital e revestidas por epitélio de transição proximalmente, mudando para epitélio escamoso simples distalmente. A porção mais distal da uretra pendular é também conhecida como fossa navicularis, igualmente derivada da placa uretral à medida que atravessa a glande do pénis, mas é composta por epitélio escamoso estratificado.
Epidemiologia
A proporção da incidência de AUV em relação à das válvulas uretrais posteriores (PUV) é de 1:8; PUV ocorre em 1 em 8000 a 1 em 25,000 nascidos vivos do sexo masculino. Divertículos da uretra anterior são incomuns, mas são a segunda forma mais comum de obstrução uretral congênita, após PUV, em lactentes e crianças. Menos de 50 casos de megalouretra haviam sido relatados na literatura até 1993, sendo o tipo escafoide mais comum. Tanto os cistos do ducto de Cowper quanto os pólipos uretrais congênitos são bastante raros.
Válvulas Uretrais Anteriores
As válvulas uretrais anteriores são a lesão obstrutiva congênita mais comum da uretra anterior, mas são 25-30 vezes menos comuns do que as PUVs. Como descrito classicamente, elas assumem a forma de uma membrana diafragmática fenestrada ou de uma cúspide mucosa originada na parede ventral da uretra. A embriologia é incerta, pois as válvulas uretrais anteriores são ocasionalmente encontradas na extremidade distal de um divertículo. Entretanto, a presença de corpo esponjoso hipoplásico sobre a porção afetada da uretra anterior sugere uma posição no espectro das hipospádias ou uma união defeituosa entre a mucosa uretral e o epitélio da fossa navicular. A ruptura de glândulas bulbouretrais dilatadas também tem sido sugerida como etiologia. Essas válvulas podem localizar-se em qualquer ponto da uretra anterior. Em 40%, está localizada na uretra bulbar, 30% na junção penoescrotal e 30% na uretra peniana. Raramente foram relatadas na fossa navicular. A apresentação geralmente é com sintomas obstrutivos. Esses pacientes comumente apresentam gotejamento de urina, dificuldade para urinar, incontinência, hesitação miccional ou retenção urinária, jato urinário fraco e infecções do trato urinário recorrentes. Crianças maiores podem também apresentar enurese, gotejamento pós-miccional ou falha de crescimento. Alterações secundárias no trato urinário superior são raras. Entretanto, a obstrução pode resultar em megacistose, ruptura vesical, hidroureteronefrose grave, lesão renal aguda ou ascite urinária no período neonatal. A cistouretrografia miccional (MCUG), a uretrografia retrógrada e a endoscopia são ferramentas diagnósticas úteis. Na MCUG, a própria válvula pode ser visualizada como um defeito linear ao longo da parede ventral, mas pode ser observada apenas como uma mudança abrupta no calibre da uretra.
O refluxo vesicoureteral foi observado em aproximadamente um terço dos pacientes, e a dilatação do trato urinário superior foi observada na metade. Endoscopicamente, a válvula se apresenta como uma cúspide ou aba de tecido fina e translúcida, localizada ventralmente, e ocasionalmente foi descrita como uma membrana semelhante à íris. Deve-se examinar minuciosamente a uretra na extremidade distal de qualquer divertículo uretral, pois o fluxo retrógrado da solução irrigante pode achatar o mecanismo valvar contra a parede uretral. A pressão suprapúbica com a bexiga cheia e as portas de irrigação do endoscópio abertas pode evidenciar o mecanismo valvar mais prontamente.
Além disso, a elevação ou a apreensão da válvula com uma alça endoscópica é inestimável para identificar a lesão. A incisão transuretral da válvula é adequada para a maioria dos pacientes, mas deve-se ter cuidado para incisar completamente, a fim de evitar qualquer lábio obstrutivo distal. O tratamento endoscópico de fato deixa o paciente com um divertículo uretral na maioria dos casos. Eventualmente, um paciente com um divertículo grande e um defeito no corpo esponjoso pode se beneficiar de correção aberta, que permite a reconstrução da válvula, do divertículo e dos tecidos envolventes. Pode ser necessária uma vesicostomia em um prematuro ou lactente pequeno para facilitar o alívio da obstrução até que o lactente possa aceitar um cistoscópio ou submeter-se a reconstrução adicional. Até 80% das crianças com válvulas uretrais anteriores desenvolverão disfunção vesical, instabilidade, hiperreflexia e redução da complacência e da capacidade observadas na urodinâmica. Devido à apresentação mais branda e sutil, a incidência global de função renal preservada em AUV é melhor do que em PUVs, com cerca de 78% dos pacientes apresentando função renal normal após o tratamento.
Divertículo uretral
Na forma de colo amplo, mais comum, geralmente localizada na região da junção penoescrotal, o lábio distal pode originar uma forma de obstrução valvular à medida que o divertículo sofre distensão progressiva. A apresentação ocorre com sintomas obstrutivos ou com gotejamento pós-miccional. As lesões saculares mais raras apresentam colo estreito e podem ocorrer em qualquer ponto ao longo de toda a uretra peniana, incluindo a fossa navicularis. Apresentam-se com infecção urinária; raramente estão associadas à formação de cálculos no interior do divertículo. O tratamento é por incisão endoscópica ou ressecção do lábio obstrutivo ou, raramente, por via perineal aberta para excisão de um divertículo grande.
Megalouretra
É uma anomalia congénita rara caracterizada por dilatação anómala da uretra peniana sem obstrução evidente. Pode estar associada à ausência do corpo esponjoso ou à ausência completa dos corpos cavernosos. Nestes casos, o pénis reduz-se a pouco mais do que um saco flácido, composto por pele externamente e mucosa uretral internamente. Partilha algumas das características de um divertículo uretral, mas inclui envolvimento mais extenso e uniforme da uretra. A classificação em tipos fusiforme e escafoide reflete a gravidade do defeito e o efeito resultante sobre o corpo esponjoso e os corpos cavernosos. A forma escafoide resulta de deficiência ou ausência do corpo esponjoso ventral. A megalouretra fusiforme também afeta o corpo esponjoso dorsal e os corpos cavernosos. Adamson e Burge descreveram um terceiro tipo com todos os corpos intactos. A causa da megalouretra permanece algo controversa. Stephens postulou que um atraso na canalização do núcleo epitelial distal poderia levar a obstrução e dilatação proximal. Outros sugeriram que uma paragem embriológica do mesoderma que reveste as pregas uretrais influencia o desenvolvimento dos corpos e do tecido erétil.
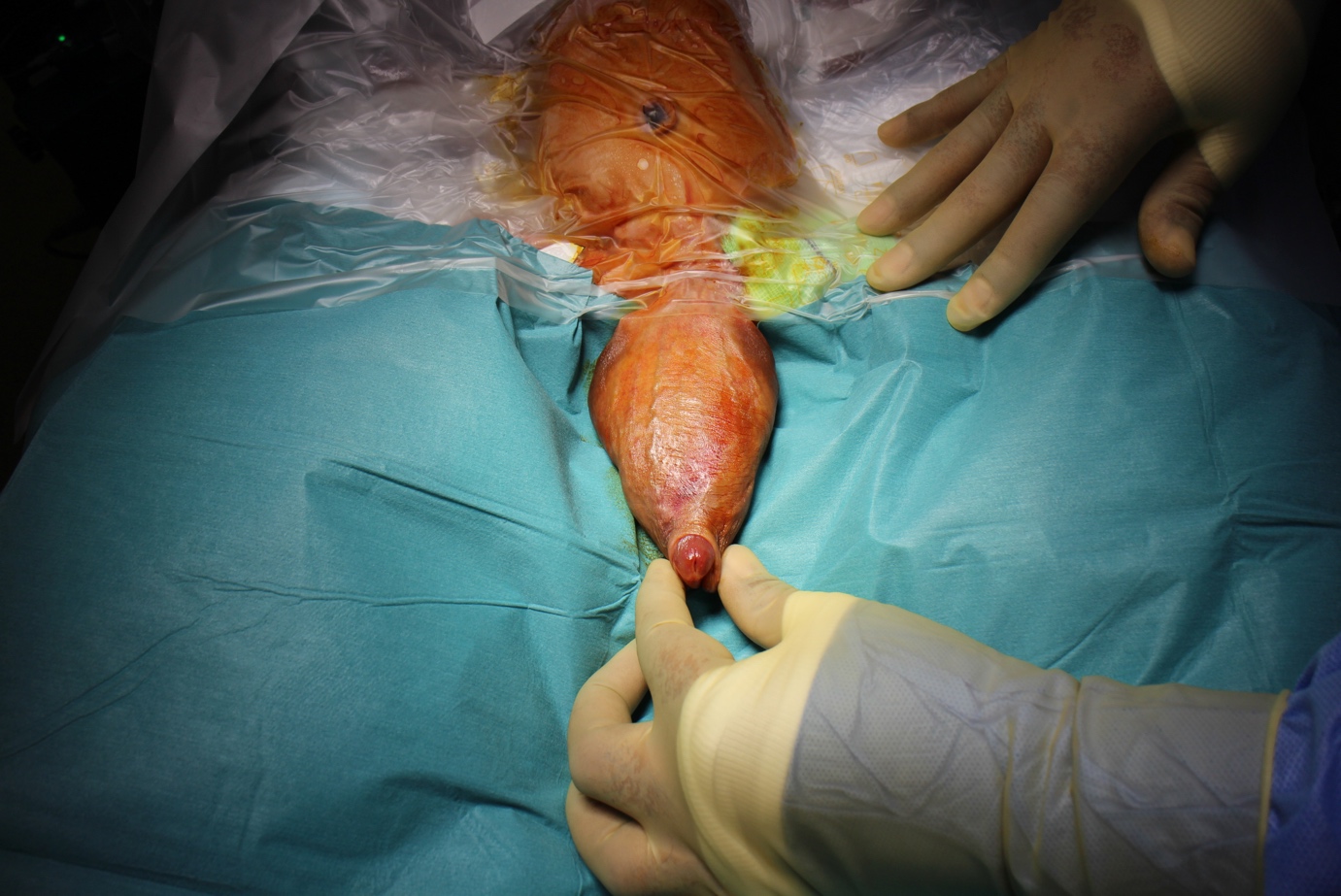
Figura 1 Exame sob anestesia mostrando grande megalouretra envolvendo a haste peniana.
Megalouretra está frequentemente associada a outras anomalias uroretais, como ânus imperfurado, síndrome de prune-belly e valvas uretrais, e a graus variados de uropatia obstrutiva. O diagnóstico é frequentemente suspeitado ao exame físico, principalmente se a criança for observada urinando. O meato uretral pode estar em posição normal, mas ser patológico. Um falo macio e alongado frequentemente caracteriza o tipo fusiforme, raro e mais grave, com corpos cavernosos palpavelmente inadequados. A dilatação ventral durante a micção é típica da megalouretra escafoide. Cistouretrografia miccional e ultrassonografia renal são realizadas para estabelecer o diagnóstico e avaliar as vias urinárias superiores. O tratamento concentra-se na reconstrução da uretra e do corpo esponjoso, utilizando princípios padrão da correção de hipospádias. A uretra escafoide pode ser aberta longitudinalmente e tubularizada utilizando o tecido dorsal e lateral de melhor qualidade. A variedade fusiforme apresenta um desafio muito mais difícil, dependendo da quantidade de tecido dos corpos cavernosos presente. Alguns casos podem ser impossíveis de reparar completamente do ponto de vista funcional e se beneficiar de reconstrução dos corpos cavernosos com próteses penianas na vida adulta.
Siringocele
Siringocele (do “syrongos”, que significa tubo, e “coele”, que significa inchaço) é uma rara dilatação cística da porção distal dos ductos das glândulas de Cowper. A glândula recebeu o nome do cirurgião e anatomista William Cowper, que a descreveu pela primeira vez no século XVII. As glândulas de Cowper são homólogas às glândulas de Bartholin nas mulheres. Essas glândulas exócrinas são glândulas bulbouretrais pares situadas de cada lado da uretra membranosa. As glândulas principais situam-se no diafragma urogenital, e as glândulas acessórias localizam-se profundamente na uretra bulbar esponjosa. Os ductos dessas glândulas atravessam o esfíncter externo e desembocam na uretra bulbar, abrindo-se separadamente ou como um único ducto, um a dois centímetros (cm) mais distalmente. As glândulas produzem secreção mucoide durante a excitação sexual e fazem parte do pré-ejaculado, o qual ajuda a lubrificar a uretra para a passagem dos espermatozoides.
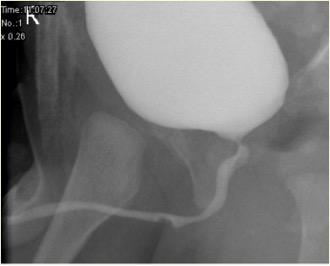
Figura 2 Aspecto de siringocele não rota na MCUG
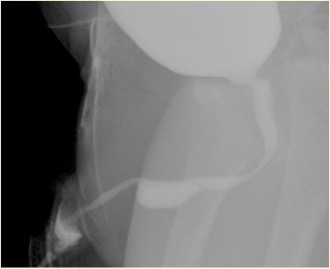
Figura 3 Aspecto de siringocele roturada na MCUG
A incidência verdadeira da siringocele é desconhecida e pode dever-se à raridade e ao desconhecimento dessa condição, agravados pela existência de diagnósticos alternativos, como um falso trajeto, divertículo uretral ou válvula uretral anterior, e, em alguns, ao potencial de resolução espontânea. As obstruções do óstio do ducto são congênitas ou adquiridas. A proliferação do epitélio ductal resulta em cistos de retenção congênitos e dilatação ductal pré-estenótica.
Infecção ou cateterismo uretral repetido são causas de obstrução adquirida. A siringocele é rara e pode se apresentar com infecções urinárias recorrentes, hematúria, sintomas obstrutivos ou gotejamento pós-miccional. Raramente pode ser um achado incidental. Obstrução significativa do trato de saída vesical devido a uma siringocele na primeira infância pode levar à doença renal crônica e à disfunção vesical. Para todos os pacientes, portanto, o diagnóstico precoce e o tratamento imediato são vitais. Uma classificação útil considera os aspectos da apresentação clínica e do manejo do paciente. Os grupos são obstrutivo, não obstrutivo sintomático e não obstrutivo assintomático. O grupo obstrutivo incluiria todos os pacientes que se apresentam com sinais ou sintomas, ou com evidências radiográficas ou endoscópicas de obstrução do trato de saída vesical. A apresentação geralmente ocorre na primeira infância.
Anomalias associadas incluem bexiga de parede espessa, rim displásico multicístico (MCDK), rim displásico e obstrução da junção pielo-ureteral (PUJ). A cistouretrografia miccional (MCUG) é um exame diagnóstico que pode mostrar um aspecto de “defeito de enchimento” da uretra (siringocele não rota) ou contraste preenchendo o cisto roto com trabeculação vesical, VUR e esvaziamento incompleto. Um exame cistoscópico pode ser confirmatório. O manejo endoscópico utiliza várias técnicas para tratar siringoceles, a saber, eletrodo de Bugbee, tesouras, cateter uretral com gancho, cateterização e Laser de Hólmio: YAG. Durante o manejo endoscópico, siringoceles não rotas são abertas, e as bordas de todas as siringoceles rotas podem ser incisadas. A siringocele não é uma condição inconsequente em lactentes, e todos os pacientes devem ser tratados adequadamente e acompanhados de perto. Na prática da urologia pediátrica, a siringocele deve ser considerada na avaliação da obstrução uretral anterior. O acompanhamento próximo de pacientes com siringocele é necessário, pois até 40% apresentam comprometimento renal, dos quais quase dois quintos progridem para insuficiência renal crônica. Uma minoria apresenta disfunção vesical.
Atresia e agenesia uretrais
A atresia e a agenesia da uretra devem ser incluídas no diagnóstico diferencial de anomalias renais e de hidronefrose bilateral diagnosticadas in útero. Infelizmente, essas lesões não são compatíveis com o desenvolvimento renal, a menos que haja alguma outra via de saída para a urina escapar da bexiga, como um úraco pérvio ou uma comunicação uroretal. O manejo dependerá da anomalia específica e da quantidade de função renal preservada pela drenagem urinária alternativa. Foi reconhecida uma associação entre atresia uretral e a síndrome de Prune-Belly em meninas, mas poucos relatos discutem o desfecho do tratamento nesses lactentes gravemente doentes. A maioria dessas crianças acabará necessitando de transplante renal devido a lesão renal grave no período neonatal. Em meninos, a dilatação progressiva com cateter tem sido utilizada de forma razoável e seria igualmente eficaz em meninas (PADUA-aumento progressivo por dilatação da uretra anterior). Ainda não está claro se isso ofereceria uma solução a longo prazo.
Duplicações uretrais
Trata-se de um conjunto raro de anomalias uretrais. Considerando as muitas variantes anatômicas diferentes, nenhuma via embriológica comum consegue explicar todas as variantes de duplicação uretral. Johnson sugeriu que uma fusão defeituosa da crista genital e da prega uretral poderia resultar em dois canais separados, e Lowsley propôs a persistência da placa urogenital durante as invaginações da crista genital como explicação. A duplicação uretral pode ocorrer com duplicação completa do falo ou da bexiga urinária como uma anomalia mais grave. Há dois tipos de duplicação: sagital ou colateral. A maioria das duplicações ocorre no plano sagital com um único falo. Quando uma é encontrada acima da outra nesse contexto, a uretra dominante desemboca em posição hipospádica e a uretra acessória na posição ortotópica. Muitas das uretras dorsais não dominantes terminam em fundo cego antes de alcançar a bexiga. Se, no entanto, alcançam a bexiga, a criança frequentemente apresenta incontinência pelo canal acessório. Na forma colateral, as uretras duplicadas dispõem-se lado a lado. A duplicação completa da bexiga e da uretra é rara, sendo particularmente rara no sexo feminino.
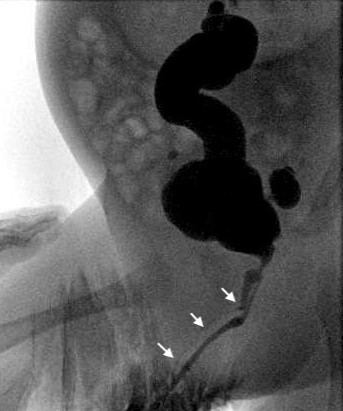
Figura 4 Cistograma demonstrando duplicação uretral em Y, com setas apontando para a uretra (dorsal) não dominante.
Em alguns casos, ambas as uretras deixam a bexiga separadamente e permanecem separadas ao longo de toda a sua extensão, ao passo que, em outros casos, as uretras duplicadas se unem distalmente para formar um único canal. Na assim chamada variedade 'fuso', a uretra se separa em dois componentes antes de se reunir novamente mais distalmente, ao passo que, nas duplicações em 'Y', uma uretra acessória diverge do canal principal para emergir na região perianal ou no períneo. Uma sínfise púbica alargada pode ser encontrada naqueles casos associados a um meato epispádico, sugerindo uma relação com o complexo extrofia–epispádias. A reconstrução cirúrgica depende da anatomia individual, mas quase sempre implica a excisão da uretra acessória mais estreita. A endoscopia no momento da reconstrução pode ocasionalmente ser necessária para compreender completamente a lesão. Se o problema for observado por causa de discreta divisão do jato urinário durante a micção, a correção pode não ser necessária. Tal consideração é tipicamente apropriada apenas para aqueles pacientes em que ambos os meatos estão imediatamente adjacentes entre si, e mesmo esses pacientes podem se beneficiar de uma simples meatoplastia para combinar ambas as aberturas em uma só. A complexidade da correção aumenta à medida que as duas aberturas divergem, especialmente se o meato ventral dominante for muito proximal. Nessa situação, a uretra perineal ventral geralmente precisa ser mobilizada e afastada do reto para uma posição mais ortotópica, e o restante da uretra reconstruído com uma variedade de técnicas de uretroplastia. Um prepúcio completo é útil quando presente. Os pacientes não devem ser submetidos à circuncisão neonatal se a anomalia for identificada. Enxertos de mucosa podem ser necessários para alguns pacientes que tenham sido submetidos previamente a circuncisão ou cirurgia. Eles também podem ser necessários para alguns pacientes cuja pele prepucial não possui comprimento adequado.
Com a dissecção agressiva necessária para reconstruir a uretra ventral, a exposição para a excisão da uretra dorsal acessória geralmente é boa. Essa estrutura também pode ser anastomosada proximalmente à uretra ventral. Passerine-Glazel e colegas descreveram a dilatação seriada gradual da uretra dorsal para torná-la útil para a micção. Preferimos a mobilização e a reconstrução da uretra dominante na maioria dos casos.
Estenose meatal
A estenose do meato uretral ocorre mais frequentemente em meninos circuncidados e é causada por dermatite da fralda. O sintoma de apresentação é geralmente um jato urinário diminuído ou desviado. A meatotomia e a meatoplastia são métodos confiáveis de tratamento.
Estenose Uretral Anterior
As estenoses da uretra anterior são frequentemente idiopáticas (talvez congênitas) ou de origem traumática. No entanto, estenoses após reparo de hipospádia também ocorrem com alguma frequência na uretra anterior; decorrentes tanto do próprio reparo quanto do cateter utilizado como stent uretral. As estenoses da uretra anterior geralmente produzem sintomas de irritação (hematúria, disúria, escapes urinários) ou de obstrução (esforço para urinar, retenção). O melhor método diagnóstico é a imagem radiográfica da uretra ou a endoscopia. A urofluxometria pode revelar um padrão de fluxo obstrutivo, mas a taxa de fluxo pode ser normal apesar da presença de uma estenose; portanto, não se pode confiar na urofluxometria para excluir uma estenose. A uretrotomia interna sob visão direta é adequada em apenas metade dos casos, mas provavelmente não é prejudicial se usada apenas uma vez. A dilatação não parece ser um tratamento apropriado, pois exige sessões repetidas muitas vezes. A anastomose uretral término-terminal após excisão da estenose é o tratamento mais eficaz quando anatomicamente factível, mesmo para estenoses pós-hipospádia. Quando a anastomose uretral não é factível, um enxerto em placa de mucosa bucal ou de pele costuma ser bem-sucedido. É melhor evitar a PDS como material de sutura no reparo uretral.
Pólipos uretrais
Pólipos uretrais são outra anomalia incomum da uretra masculina. Podem ser congênitos, mas também foram relatados em adultos, sugerindo que podem ser adquiridos ou crescer lentamente até se tornarem grandes o suficiente para causar sintomas. Essas lesões fibroendoteliais comumente surgem do verumontanum e são tipicamente cobertas por epitélio de transição sobre um núcleo fibromuscular. Pólipos pequenos geralmente são descobertos incidentalmente durante endoscopia por algum motivo não relacionado. Em contraste, as lesões mais extensas, com uma cabeça polipoide flutuando livremente em um pedículo alongado, tendem a protruir através do colo vesical, dando origem a episódios agudos e transitórios de retenção urinária. Outros sintomas são esforço miccional, urgência e hematúria. Pode também ocorrer hemorragia uretral franca. O diagnóstico é feito por MCU ou cistouretroscopia. A cistouretrografia miccional frequentemente mostra um defeito de enchimento na uretra, que pode variar de localização. A confirmação diagnóstica é feita por cistoscopia. A maioria dos pólipos pode ser excisada endoscopicamente (excisão transuretral), embora, para pólipos grandes, possa ser necessária uma abordagem transvesical aberta.
Pontos-chave
- Válvulas uretrais anteriores são a lesão obstrutiva congênita mais comum da uretra anterior e geralmente se apresentam com sintomas obstrutivos. A incisão transuretral da válvula é adequada para a maioria dos pacientes.
- Divertículo uretral pode se apresentar na forma mais comum de boca ampla ou na forma sacular. O tratamento é por incisão endoscópica ou ressecção do lábio obstrutivo ou, raramente, por uma abordagem perineal aberta, com excisão de um grande divertículo.
- Megalouretra É uma anomalia congênita rara caracterizada por dilatação anormal da uretra peniana sem obstrução estrutural. Cistouretrografia miccional e ultrassonografia renal são realizadas para estabelecer o diagnóstico e avaliar as vias urinárias superiores. O tratamento centra-se na reconstrução da uretra e do corpo esponjoso usando princípios padrão da correção da hipospádia.
- Siringocele é uma dilatação cística rara da porção distal dos ductos das glândulas de Cowper. Para todos os pacientes, diagnóstico precoce e tratamento imediato são vitais. O manejo endoscópico utiliza várias técnicas para tratar siringoceles, a saber, eletrodo Bugbee, tesouras, cateter uretral com gancho, cateterização e Laser de Hólmio: YAG.
- Atresia e agenesia uretrais devem ser incluídas no diagnóstico diferencial de anomalias renais e hidronefrose bilateral diagnosticadas intraútero. O manejo dependerá da anomalia específica e da quantidade de função renal preservada por drenagem urinária alternativa. Em pacientes do sexo masculino, a dilatação progressiva com cateter tem sido utilizada de maneira razoável e seria igualmente eficaz em pacientes do sexo feminino (PADUA-aumento progressivo pela dilatação da uretra anterior).
- A duplicação uretral é um espectro raro de anomalias uretrais. A complexidade da correção aumenta à medida que as duas aberturas se separam, especialmente se o meato ventral dominante for muito proximal.
- Estenose meatal ocorre com mais frequência em meninos circuncidados e é causada por dermatite de fralda. Meatotomia e meatoplastia são métodos de manejo confiáveis.
- Estenoses uretrais anteriores da uretra anterior são frequentemente idiopáticas (talvez congênitas) ou de origem traumática. A anastomose uretral término-terminal após excisão da estenose é o tratamento mais eficaz quando anatomicamente factível, mesmo para estenoses pós-hipospádia.
- Pólipos uretrais são uma anomalia incomum da uretra masculina. Essas lesões fibro-endoteliais comumente se originam do verumontanum. O diagnóstico é por MCU ou cistouretroscopia. A maioria dos pólipos pode ser excisada endoscopicamente (excisão transuretral), embora, para pólipos grandes, possa ser necessária uma abordagem transvesical aberta.
Referências
- Antón-Juanilla M, Lozano-Ortega JL, Galbarriatu-Gutiérrez A. A Urresola-Olabarrieta, Anterior urethral trauma in childhood: presentation of two cases. Cir Pediatr 2020; 1;33(4):200-203.
- Ansari MS, Yadav P, Srivastava A, Kapoor R, Shekar PA. Etiology, and characteristics of pediatric urethral strictures in a developing country in the 21st century. J Pediatr Urol 2019; 15: 403.e1–403.e8. DOI: 10.1016/j.jpurol.2019.05.020.
- Vetterlein MW, Weisbach L, Reichardt S, Fisch M. Anterior Urethral Strictures in Children: Disease Etiology and Comparative Effectiveness of Endoscopic Treatment vs Open Surgical Reconstruction. Pediatr 2019. DOI: 10.3389/fped.2019.00005.
- Kaplan GW, Brock JW, Fisch M, Koraitim MM, Snyder HM. SIU/ICUD Consultation on Urethral Strictures: Urethral Strictures in Children. Urology 2014; 83. DOI: 10.1016/j.urology.2013.09.010.
- Perlman S, Borovitz Y, Ben-Meir D, Hazan Y, Bardin RNR, Brusilov M, et al.. Prenatal diagnosis and postnatal outcome of anterior urethral anomalies. 2020; 40 (2): 191–196. DOI: 10.1002/pd.5582.
- Thomas D, Duffy P, Rickwood A. Essentials of Paediatric Urology. 2008: 117–120. DOI: 10.1046/j.1464-410x.2002.t01-1-03073_1.x.
- Gillenwater J, Howards S, Grayhack J, Mitchell M. Adult and Pediatric Urology ". 4th ed., USA: Lippincott Williams & Wilkins Publishers; 2002, DOI: 10.1001/jama.1991.03460240115044.
- Stringer M, Oldham K, Mouriquand P. Pediatric Surgery and Urology: long – term outcomes". 2nd ed., USA: Cambridge University Press; 2006, DOI: 10.1016/s0002-9610(99)00082-3.
- Berrocal T, López-Pereira P, Arjonilla A, Gutiérrez J. Anomalies of the distal ureter, bladder, and urethra in children: embryologic, radiologic and pathologic features". Radiographics 2002. DOI: 10.1148/radiographics.22.5.g02se101139.
- Alonso AR, Outeda EC, Blanco AG, Martín CB, Franco JL, Pérez MAC, et al.. Duplicidad de uretra masculina. Actas Urológicas Españolas 2002; 26 (1): 69–73.
Ultima atualização: 2025-09-21 13:35