
25: Anomalías por duplicación uretral y siringocele
Este capítulo durará aproximadamente 15 minutos para leer.
Introducción
Las anomalías uretrales son trastornos congénitos que no son infrecuentes, con algunas variantes raras. Este capítulo se centrará en la embriología, epidemiología, diagnóstico, evaluación, reparación, seguimiento y complicaciones de algunas anomalías uretrales anteriores, incluyendo la válvula uretral anterior (VUA), el siringocele, la duplicación uretral y la megalouretra. Algunos incluso creen que estas anomalías, excepto la duplicación, constituyen una misma entidad y un espectro.
Embriología
El desarrollo de la uretra masculina se origina cuando la cavidad del seno urogenital se extiende hacia la superficie del tubérculo genital durante la semana 6 de gestación. Este surco de origen endodérmico se convierte en una placa sólida de células, que eventualmente se tubulariza en sentido proximal a distal para formar la uretra fálica. La apariencia es idéntica en fetos masculinos y femeninos hasta la semana nueve, y para la semana 14 la uretra masculina ha completado su desarrollo. La uretra se ha dividido en cuatro regiones distintas, cada una con su clasificación anatómica única y con un tipo celular histológico predominante de la mucosa uretral. La uretra prostática y la uretra membranosa son las más proximales, derivadas del seno urogenital y compuestas por un delicado epitelio de transición. A continuación se encuentran la uretra bulbar y la uretra pendular, derivadas de la placa uretral en la cara ventral del tubérculo genital y revestidas por epitelio de transición en la porción proximal, cambiando a epitelio escamoso simple distalmente. La porción más distal de la uretra pendular también se conoce como fosa navicular, igualmente derivada de la placa uretral a medida que atraviesa el glande del pene, pero compuesta por epitelio escamoso estratificado.
Epidemiología
La proporción de la incidencia de AUV con respecto a la de las válvulas uretrales posteriores (PUV) es 1:8; las PUV ocurren en 1 de cada 8000 a 1 de cada 25,000 nacidos vivos varones. Los divertículos de la uretra anterior son poco frecuentes, pero constituyen la segunda forma más común de obstrucción uretral congénita, después de las PUV, en lactantes y niños. Para 1993, se habían reportado en la literatura menos de 50 casos de megalouretra, siendo más común el tipo escafoide. Tanto los quistes del conducto de Cowper como los pólipos uretrales congénitos son bastante raros.
Válvulas uretrales anteriores
Las válvulas uretrales anteriores son la lesión obstructiva congénita más común de la uretra anterior, pero son 25-30 veces menos frecuentes que las PUVs. Como se describe clásicamente, adoptan la forma de una membrana diafragmática fenestrada o de una cúspide mucosa que se origina en la pared ventral de la uretra. La embriología no está clara porque las válvulas uretrales anteriores se encuentran ocasionalmente en el extremo distal de un divertículo. Sin embargo, un cuerpo esponjoso hipoplásico sobre la porción afectada de la uretra anterior indica una posición dentro del espectro de hipospadias o una unión defectuosa entre la mucosa uretral y el epitelio de la fosa navicular. También se ha sugerido, como etiología, la ruptura de glándulas bulbouretrales dilatadas. Estas válvulas pueden ubicarse en cualquier parte de la uretra anterior. En el 40%, se sitúan en la uretra bulbar; en el 30%, en la unión penoescrotal; y en el 30%, en la uretra peneana. Rara vez se han reportado en la fosa navicular. La presentación suele ser con síntomas obstructivos. Estos pacientes suelen presentar goteo urinario, dificultad miccional, incontinencia, vacilación miccional o retención urinaria, chorro urinario débil e infecciones urinarias recurrentes. Los niños mayores también pueden presentar enuresis, goteo postmiccional o falta de medro. Los cambios secundarios en el tracto urinario superior son raros. Sin embargo, la obstrucción puede producir megacistis, rotura vesical, hidroureteronefrosis grave, lesión renal aguda o ascitis urinaria en el periodo neonatal. La cistouretrografía miccional (MCUG), la uretrografía retrógrada y la endoscopia son herramientas diagnósticas útiles. En la MCUG, la propia válvula puede visualizarse como un defecto lineal a lo largo de la pared ventral, pero puede apreciarse únicamente como un cambio brusco en el calibre de la uretra.
Se ha observado reflujo vesicoureteral en aproximadamente un tercio de los pacientes, y dilatación de las vías urinarias superiores en la mitad. Endoscópicamente, la válvula aparece como una cúspide o colgajo de tejido fino y membranoso, ubicado ventralmente, y ocasionalmente se ha descrito como una membrana en forma de iris. Debe examinarse minuciosamente la uretra en el extremo distal de cualquier divertículo uretral porque el flujo retrógrado del irrigante puede aplanar el mecanismo valvular contra la pared uretral. La presión suprapúbica con la vejiga llena y los puertos de irrigación del endoscopio abiertos puede demostrar el mecanismo valvular con mayor facilidad.
Además, la elevación o el enganche de la válvula con un asa endoscópica es inestimable para identificar la lesión. La incisión transuretral de la válvula es suficiente para la mayoría de los pacientes, pero se debe tener cuidado de incidir por completo para evitar cualquier reborde obstructivo distal. El tratamiento endoscópico sí deja al paciente con un divertículo uretral en la mayoría de los casos. De forma ocasional, un paciente con un divertículo grande y un defecto del cuerpo esponjoso puede beneficiarse de una reparación abierta, que permite la reconstrucción de la válvula, el divertículo y los tejidos circundantes. Puede ser necesaria una vesicostomía en un lactante prematuro o pequeño para facilitar el alivio de la obstrucción hasta que el lactante pueda permitir el paso de un cistoscopio o someterse a una reconstrucción adicional. Hasta el 80% de los niños con válvulas uretrales anteriores desarrollarán disfunción vesical, inestabilidad, hiperreflexia y disminución de la distensibilidad y la capacidad evidenciadas en urodinamia. Debido a la presentación más leve y sutil, la incidencia global de función renal preservada en las VUA es mejor que en las VUP, con alrededor del 78% de los pacientes presentando función renal normal tras el tratamiento.
Divertículo uretral
En la forma más común de boca ancha, por lo general localizada en la región de la unión penoescrotal, el labio distal puede dar lugar a una forma de obstrucción valvular a medida que el divertículo sufre una distensión progresiva. La presentación es ya sea con síntomas obstructivos o con goteo posmiccional. Las lesiones saculares más raras tienen un cuello estrecho y pueden aparecer en cualquier punto a lo largo de la uretra peneana, incluida la fosa navicular. Se presentan con infección urinaria; en raras ocasiones se asocian con la formación de cálculos dentro del divertículo. El tratamiento consiste en incisión endoscópica o resección del labio obstructivo o, en raras ocasiones, en un abordaje perineal abierto para extirpar un divertículo grande.
Megalouretra
Es una anomalía congénita rara caracterizada por una dilatación anómala de la uretra peneana sin obstrucción clara. Puede asociarse con ausencia del cuerpo esponjoso o con la ausencia completa de los cuerpos cavernosos. En tales casos, el pene se reduce a poco más que un saco flácido compuesto externamente por piel e internamente por mucosa uretral. Comparte algunas de las características de un divertículo uretral, pero incluye una afectación más extensa y uniforme de la uretra. La clasificación en tipos fusiforme y escafoide refleja la gravedad del defecto y el efecto resultante sobre el cuerpo esponjoso y los cuerpos cavernosos. La forma escafoide resulta de la deficiencia o ausencia del cuerpo esponjoso ventral. Una megalouretra fusiforme también afecta el cuerpo esponjoso dorsal y los cuerpos cavernosos. Adamson y Burge han descrito un tercer tipo con todos los cuerpos íntegros. La causa de la megalouretra sigue siendo algo controvertida. Stephens postuló que un retraso en la canalización del núcleo epitelial distal podría llevar a obstrucción y dilatación proximal. Otros han sugerido que el arresto embriológico del mesodermo que reviste los pliegues uretrales influye en el desarrollo de los cuerpos y del tejido eréctil.
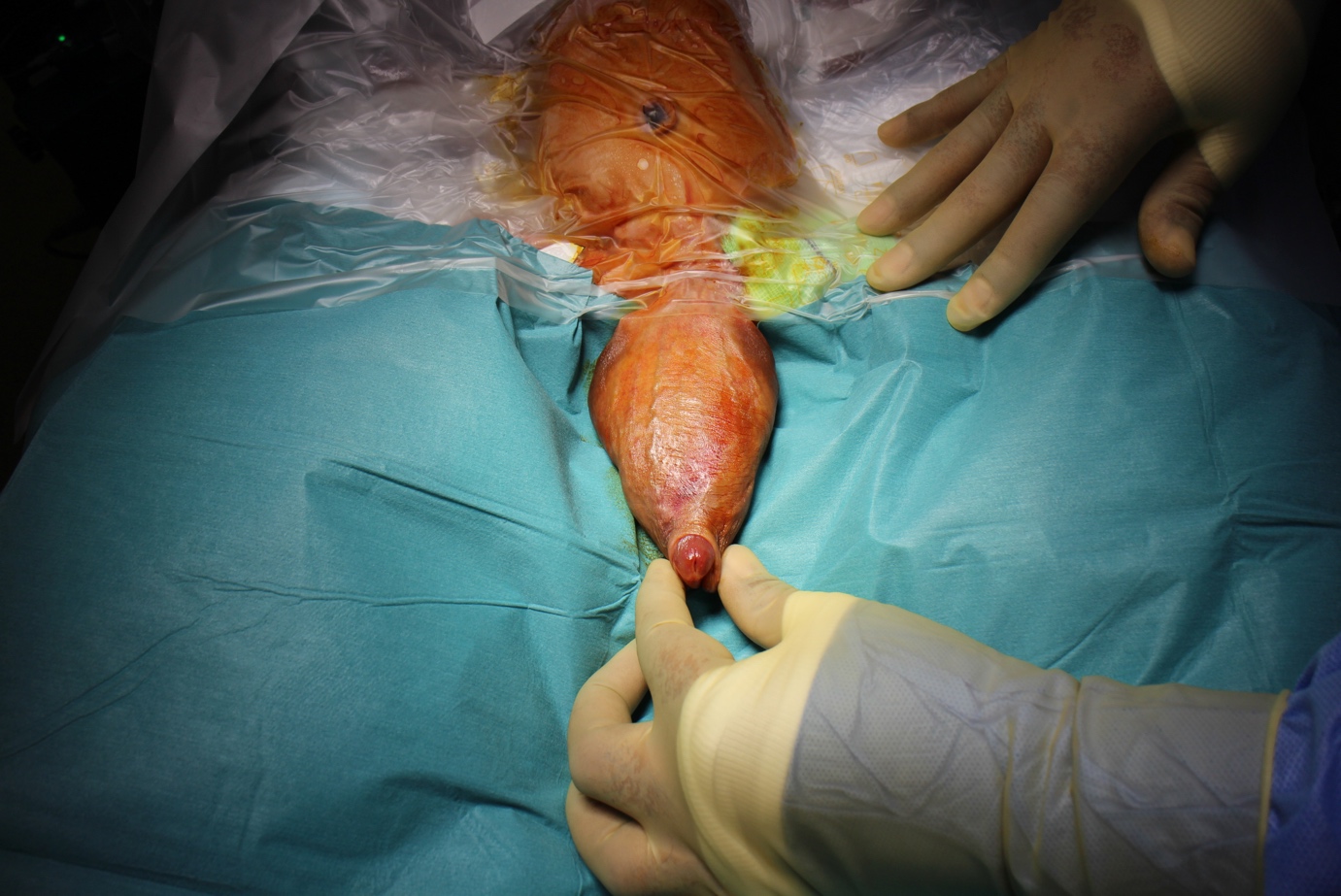
Figura 1 Exploración bajo anestesia que muestra una megalouretra de gran tamaño que compromete el cuerpo del pene.
La megalouretra suele asociarse con otras anomalías urorectales como el ano imperforado, el síndrome de abdomen en ciruela pasa y las válvulas uretrales, y con grados variables de uropatía obstructiva. El diagnóstico suele sospecharse en el examen físico, sobre todo si se le observa orinar al niño. El meato uretral puede estar en posición normal pero ser patológico. Un falo blando y alargado suele caracterizar el tipo fusiforme, raro y más grave, con cuerpos cavernosos palpablemente insuficientes. La distensión ventral durante la micción es típica de la megalouretra escafoide. La cistouretrografía miccional y la ecografía renal se realizan para establecer el diagnóstico y evaluar las vías urinarias superiores. El tratamiento se centra en reconstruir la uretra y el cuerpo esponjoso utilizando principios estándar de la reparación del hipospadias. La uretra escafoide puede abrirse longitudinalmente y tubularizarse utilizando el mejor tejido dorsal y lateral. La variedad fusiforme plantea un desafío mucho más difícil, según la cantidad de tejido de los cuerpos eréctiles presente. Algunos pueden ser imposibles de reparar por completo desde el punto de vista funcional y beneficiarse en la edad adulta de la reconstrucción de los cuerpos cavernosos con prótesis peneanas.
Siringocele
Siringocele (del “syrongos”, que significa tubo, y “coele”, que significa tumefacción) es una rara dilatación quística de la porción distal de los conductos de la glándula de Cowper. La glándula recibe su nombre del cirujano y anatomista William Cowper, quien la describió por primera vez en el siglo XVII. Las glándulas de Cowper son homólogas a las glándulas de Bartolino en las mujeres. Estas glándulas exocrinas son glándulas bulbouretrales pares situadas a cada lado de la uretra membranosa. Las glándulas principales se sitúan sobre el diafragma urogenital, y las glándulas accesorias se encuentran profundamente dentro de la uretra bulbar esponjosa. Los conductos de estas glándulas atraviesan el esfínter externo y desembocan en la uretra bulbar, abriéndose por separado o como un único conducto unido uno a dos centímetros (cm) más distalmente. Las glándulas producen una secreción mucoide durante la excitación sexual y forman parte del preeyaculado que ayuda a lubricar la uretra para el paso de los espermatozoides.
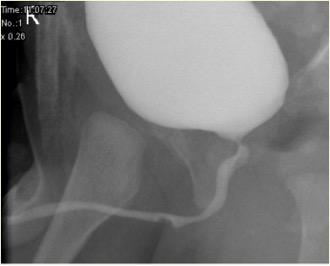
Figura 2 Aspecto del siringocele no roto en MCUG
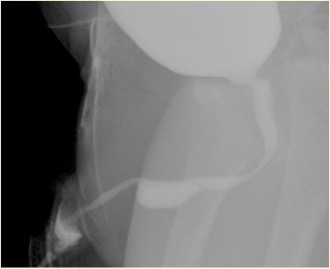
Figura 3 Aspecto de siringocele roto en la MCUG
La verdadera incidencia del siringocele es desconocida y podría deberse a la rareza y el desconocimiento de esta afección, agravados por la existencia de diagnósticos alternativos como una falsa vía, un divertículo uretral o una válvula uretral anterior y, en algunos casos, a la posibilidad de resolución espontánea. Las obstrucciones del orificio del conducto son congénitas o adquiridas. La proliferación del epitelio ductal da lugar a quistes de retención congénitos y a dilatación ductal preestenótica.
La infección o la cateterización uretral repetida son causas de obstrucción adquirida. El siringocele es poco frecuente y puede presentarse con infecciones urinarias recurrentes, hematuria, síntomas obstructivos o goteo postmiccional. Rara vez puede ser un hallazgo incidental. Una obstrucción significativa de la salida vesical debida a un siringocele en etapas tempranas de la vida puede conducir a enfermedad renal crónica y disfunción vesical. Por lo tanto, en todos los pacientes, el diagnóstico precoz y el tratamiento oportuno son fundamentales. Una clasificación útil contempla los aspectos de la presentación clínica y del manejo del paciente. Los grupos son: obstructivo, no obstructivo sintomático y no obstructivo asintomático. El grupo obstructivo incluiría a todos los pacientes que presenten signos o síntomas, o evidencia radiográfica o endoscópica de obstrucción de la salida vesical. La presentación suele darse en los primeros meses de vida.
Las anomalías asociadas incluyen vejiga con pared engrosada, riñón displásico multiquístico (MCDK), riñón displásico y obstrucción de la unión pieloureteral (PUJ). La cistouretrografía miccional (MCUG) es una prueba diagnóstica que puede mostrar un aspecto de “defecto de repleción” de la uretra (siringocele no roto) o el llenado por contraste del quiste roto con trabeculación vesical, VUR y vaciamiento incompleto. Una exploración cistoscópica puede ser confirmatoria. El manejo endoscópico utiliza diversas técnicas para tratar los siringoceles, a saber, electrodo de Bugbee, tijeras, catéter uretral con gancho, cateterización y Láser de holmio: YAG. Durante el manejo endoscópico, los siringoceles no rotos se abren y se pueden incidir los bordes de todos los siringoceles rotos. El siringocele no es una afección intrascendente en lactantes y todos los pacientes deben ser tratados adecuadamente y seguidos de cerca. En la práctica de la urología pediátrica, debe considerarse el siringocele al evaluar la obstrucción uretral anterior. Es necesario un seguimiento estrecho de los pacientes con siringocele, ya que hasta el 40% presentan deterioro renal, de los cuales casi dos quintos progresan a insuficiencia renal crónica. Una minoría presenta disfunción vesical.
Atresia y agenesia uretrales
La atresia y la agenesia uretrales deben incluirse en el diagnóstico diferencial de las anomalías renales y de la hidronefrosis bilateral diagnosticadas in útero. Lamentablemente, estas lesiones no son compatibles con el desarrollo renal a menos que exista alguna otra vía de salida para que la orina escape de la vejiga, como un uraco permeable o una comunicación urorectal. El manejo dependerá de la anomalía específica y de la cantidad de función renal que se logre preservar mediante un drenaje urinario alternativo. Se ha reconocido una asociación entre la atresia uretral y el síndrome de prune-belly en niñas, pero pocos informes abordan el resultado del tratamiento en estos lactantes gravemente enfermos. La mayoría de estos niños terminarán requiriendo trasplante renal debido al daño renal grave en el periodo neonatal. En varones, se ha utilizado con resultados razonables la dilatación progresiva con catéter y sería igualmente eficaz en niñas (PADUA-aumento progresivo mediante dilatación de la uretra anterior). Aún no está claro si esto ofrecería una solución a largo plazo.
Duplicaciones uretrales
Es un conjunto poco frecuente de anomalías uretrales. Dadas las numerosas variantes anatómicas, no existe una vía embriológica común que pueda explicar todas las variantes de duplicación uretral. Johnson sugirió que una fusión defectuosa de la cresta genital y el pliegue uretral podría dar lugar a dos conductos separados, y Lowsley propuso como explicación la persistencia de la placa urogenital durante las invaginaciones de la cresta genital. La duplicación uretral puede ocurrir con duplicación completa del falo o de la vejiga urinaria como una anomalía más grave. Hay dos tipos de duplicación, ya sea sagital o colateral. La mayoría de las duplicaciones ocurren en el plano sagital con un solo falo. Cuando una se encuentra por encima de la otra en este contexto, la uretra dominante desemboca en una posición hipospádica y la uretra accesoria en la posición ortotópica. Muchas de las uretras dorsales no dominantes terminan ciegamente antes de la vejiga. Si, sin embargo, llegan a la vejiga, el niño suele ser incontinente a través del conducto accesorio. En la forma colateral, las uretras duplicadas discurren lado a lado. La duplicación completa de la vejiga y la uretra es rara y particularmente en mujeres.
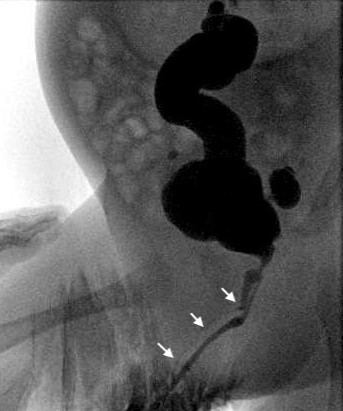
Figura 4 Estudio cistográfico que muestra duplicación uretral en Y con flechas que señalan la uretra no dominante (dorsal).
En algunos casos, ambas uretras salen de la vejiga por separado y permanecen separadas a lo largo de toda su longitud, mientras que en otros casos las uretras duplicadas se unen distalmente para formar un único conducto. En la denominada variedad 'huso', la uretra se separa en dos componentes antes de reunirse de nuevo más distalmente, mientras que, en las duplicaciones en 'Y', una uretra accesoria se desvía del conducto principal para emerger en la región perianal o el periné. Puede encontrarse una sínfisis púbica ensanchada en aquellos casos asociados a un meato epispádico, lo que sugiere una relación con el complejo de extrofia–epispadias. La reconstrucción quirúrgica depende de la anatomía individual, pero casi siempre conlleva la extirpación de la uretra accesoria más estrecha. Puede ser necesaria ocasionalmente la endoscopia en el momento de la reconstrucción para comprender a fondo la lesión. Si el problema se advierte por una división leve del chorro urinario durante la micción, puede no ser necesaria la reparación. Dicha consideración suele ser apropiada únicamente para aquellos pacientes con ambos meatos inmediatamente adyacentes entre sí, e incluso esos pacientes pueden beneficiarse de una meatoplastia simple para combinar ambas aberturas en una sola. La complejidad de la reparación aumenta a medida que ambas aberturas divergen, especialmente si el meato ventral dominante es muy proximal. En esa situación, por lo general la uretra perineal ventral debe movilizarse alejándola del recto hacia una posición más ortotópica, y el resto de la uretra se reconstruye con diversas técnicas de uretroplastia. Un prepucio completo es útil cuando está presente. Los pacientes no deben someterse a circuncisión neonatal si se observa la anomalía. Pueden ser necesarios injertos mucosos en algunos pacientes que han sido sometidos previamente a circuncisión o cirugía. También pueden ser necesarios en algunos pacientes cuya piel prepucial no tiene una longitud adecuada.
Con la disección agresiva necesaria para reconstruir la uretra ventral, la exposición para la exéresis de la uretra dorsal accesoria suele ser buena. Esa estructura también puede anastomosarse proximalmente a la uretra ventral. Passerine-Glazel y colegas han descrito la dilatación seriada gradual de la uretra dorsal para hacerla útil para la micción. Preferimos la movilización y reconstrucción de la uretra dominante en la mayoría de los casos.
Estenosis meatal
La estenosis meatal se presenta con mayor frecuencia en niños circuncidados y se debe a dermatitis del pañal. El síntoma de presentación suele ser un chorro urinario disminuido o desviado. La meatotomía y la meatoplastia son métodos fiables de manejo.
Estenosis uretral anterior
Las estenosis de la uretra anterior suelen ser idiopáticas (quizás congénitas) o de origen traumático. Sin embargo, también se presentan con cierta frecuencia estenosis posteriores a la reparación de hipospadias en la uretra anterior; como consecuencia ya sea de la propia reparación o del catéter utilizado como tutor uretral. Las estenosis de la uretra anterior suelen producir síntomas de irritación (hematuria, disuria, incontinencia urinaria) o de obstrucción (esfuerzo para orinar, retención urinaria). Se diagnostican mejor con imágenes radiográficas de la uretra o con endoscopia. La uroflujometría puede revelar un patrón de flujo obstructivo, pero la velocidad de flujo puede ser normal a pesar de la presencia de una estenosis; por lo tanto, no se puede confiar en la uroflujometría para descartar una estenosis. La uretrotomía interna a visión directa es adecuada solo en la mitad de los casos, pero probablemente no sea perjudicial si se utiliza una sola vez. La dilatación no parece ser un tratamiento adecuado, ya que requiere múltiples sesiones repetidas. La anastomosis uretral término-terminal tras la exéresis de la estenosis es el tratamiento más eficaz cuando es anatómicamente factible, incluso para las estenosis posreparación de hipospadias. Cuando la anastomosis uretral no es factible, un injerto en parche de mucosa bucal o de piel suele ser exitoso. Es preferible evitar el PDS como material de sutura en la reparación uretral.
Pólipos uretrales
Los pólipos uretrales son otra anomalía inusual de la uretra masculina. Pueden ser congénitos, pero también se han descrito en adultos, lo que sugiere que pueden ser adquiridos o crecer lentamente hasta alcanzar un tamaño suficiente para causar síntomas. Estas lesiones fibroendoteliales suelen originarse en el verumontanum y están típicamente cubiertas por epitelio transicional sobre un núcleo fibromuscular. Los pólipos pequeños suelen descubrirse de manera incidental durante una endoscopia realizada por algún motivo no relacionado. En contraste, las lesiones más extensas, con una cabeza polipoide que flota libremente sobre un pedículo alargado, tienden a protruir a través del cuello vesical y dar lugar a episodios agudos y transitorios de retención urinaria. Otros síntomas son esfuerzo miccional, urgencia y hematuria. También puede presentarse sangrado uretral franco. El diagnóstico es mediante MCU o cistouretroscopia. La cistouretrografía miccional a menudo muestra un defecto de llenado en la uretra que puede variar en su localización. La confirmación diagnóstica se realiza por cistoscopia. La mayoría de los pólipos pueden extirparse endoscópicamente (escisión transuretral), aunque, en pólipos grandes, puede requerirse un abordaje transvesical abierto.
Puntos clave
- Las válvulas uretrales anteriores son la lesión obstructiva congénita más común de la uretra anterior y suelen presentarse con síntomas obstructivos. La incisión transuretral de la válvula es adecuada para la mayoría de los pacientes.
- El divertículo uretral puede presentarse en la forma más común de boca ancha o en forma sacular. El tratamiento se realiza mediante incisión endoscópica o resección del reborde obstructivo o, raramente, mediante un abordaje perineal abierto con exéresis de un divertículo grande.
- Megalouretra Es una rara anomalía congénita caracterizada por dilatación anormal de la uretra peneana sin una obstrucción estructural. La cistouretrografía miccional y la ecografía renal se realizan para establecer el diagnóstico y evaluar las vías urinarias superiores. El tratamiento se centra en reconstruir la uretra y el cuerpo esponjoso utilizando principios estándar de la reparación del hipospadias.
- Siringocele es una dilatación quística rara de la porción distal de los conductos de la glándula de Cowper. Para todos los pacientes, el diagnóstico precoz y el tratamiento oportuno son vitales. El manejo endoscópico utiliza varias técnicas para tratar los siringoceles, a saber: electrodo Bugbee, tijeras, catéter uretral con gancho, cateterización y láser de holmio:YAG.
- La atresia y la agenesia uretrales deben incluirse en el diagnóstico diferencial de anomalías renales e hidronefrosis bilateral diagnosticadas in útero. El manejo dependerá de la anomalía específica y de la cantidad de función renal rescatada mediante drenaje urinario alternativo. En varones, la dilatación progresiva con catéter se ha utilizado razonablemente y sería de eficacia similar en mujeres (PADUA-aumento progresivo mediante dilatación de la uretra anterior).
- La duplicación uretral es un espectro raro de anomalías uretrales. La complejidad de la reparación aumenta a medida que las dos aberturas divergen, especialmente si el meato ventral dominante es muy proximal.
- La estenosis meatal aparece con mayor frecuencia en niños circuncidados y es causada por dermatitis del pañal. La meatotomía y la meatoplastia son métodos de manejo fiables.
- Las estenosis de la uretra anterior son a menudo idiopáticas (quizá congénitas) o de origen traumático. La anastomosis uretral término-terminal tras la exéresis de la estenosis es el tratamiento más eficaz cuando es anatómicamente factible, incluso para estenosis poshipospadias.
- Los pólipos uretrales son una anomalía poco frecuente de la uretra masculina. Estas lesiones fibroendoteliales suelen originarse en el verumontanum. El diagnóstico se realiza mediante MCU o cistouretroscopia. La mayoría de los pólipos pueden extirparse endoscópicamente (exéresis transuretral), aunque, para pólipos grandes, puede requerirse un abordaje transvesical abierto.
Referencias
- Antón-Juanilla M, Lozano-Ortega JL, Galbarriatu-Gutiérrez A. A Urresola-Olabarrieta, Anterior urethral trauma in childhood: presentation of two cases. Cir Pediatr 2020; 1;33(4):200-203.
- Ansari MS, Yadav P, Srivastava A, Kapoor R, Shekar PA. Etiology, and characteristics of pediatric urethral strictures in a developing country in the 21st century. J Pediatr Urol 2019; 15: 403.e1–403.e8. DOI: 10.1016/j.jpurol.2019.05.020.
- Vetterlein MW, Weisbach L, Reichardt S, Fisch M. Anterior Urethral Strictures in Children: Disease Etiology and Comparative Effectiveness of Endoscopic Treatment vs Open Surgical Reconstruction. Pediatr 2019. DOI: 10.3389/fped.2019.00005.
- Kaplan GW, Brock JW, Fisch M, Koraitim MM, Snyder HM. SIU/ICUD Consultation on Urethral Strictures: Urethral Strictures in Children. Urology 2014; 83. DOI: 10.1016/j.urology.2013.09.010.
- Perlman S, Borovitz Y, Ben-Meir D, Hazan Y, Bardin RNR, Brusilov M, et al.. Prenatal diagnosis and postnatal outcome of anterior urethral anomalies. 2020; 40 (2): 191–196. DOI: 10.1002/pd.5582.
- Thomas D, Duffy P, Rickwood A. Essentials of Paediatric Urology. 2008: 117–120. DOI: 10.1046/j.1464-410x.2002.t01-1-03073_1.x.
- Gillenwater J, Howards S, Grayhack J, Mitchell M. Adult and Pediatric Urology ". 4th ed., USA: Lippincott Williams & Wilkins Publishers; 2002, DOI: 10.1001/jama.1991.03460240115044.
- Stringer M, Oldham K, Mouriquand P. Pediatric Surgery and Urology: long – term outcomes". 2nd ed., USA: Cambridge University Press; 2006, DOI: 10.1016/s0002-9610(99)00082-3.
- Berrocal T, López-Pereira P, Arjonilla A, Gutiérrez J. Anomalies of the distal ureter, bladder, and urethra in children: embryologic, radiologic and pathologic features". Radiographics 2002. DOI: 10.1148/radiographics.22.5.g02se101139.
- Alonso AR, Outeda EC, Blanco AG, Martín CB, Franco JL, Pérez MAC, et al.. Duplicidad de uretra masculina. Actas Urológicas Españolas 2002; 26 (1): 69–73.
Última actualización: 2025-09-21 13:35