
25: 尿道重复畸形与库珀腺导管囊肿
阅读本章大约需要 3 分钟。
引言
尿道畸形是并不少见的先天性异常,但也有一些罕见的变异。本章将重点讨论一些前尿道畸形的胚胎学、流行病学、诊断、评估、修复、随访及并发症,包括前尿道瓣膜 (AUV)、球尿道腺管囊肿 (syringocele)、尿道重复和巨大尿道 (megalourethra)。甚至有人认为,除尿道重复外,这些异常本质相同,构成同一疾病谱。
胚胎学
男性尿道的发育起始于妊娠第6周,尿生殖窦腔延伸至生殖结节表面。这个来源于内胚层的沟最终变为实性细胞板,随后自近端向远端发生管状化,形成阴茎部尿道。在第9周之前,男女胎儿的外观相同;至第14周,男性尿道的发育即告完成。尿道被划分为四个不同的区域,每个区域在解剖学分类以及尿道黏膜主要的组织学细胞类型方面各具特点。最接近近端的是前列腺部尿道和膜部尿道,源自尿生殖窦,由细薄的移行上皮构成。其后为球部和悬垂部尿道,来源于生殖结节腹侧的尿道板,近端为移行上皮,向远端转变为单层鳞状上皮。最远端的悬垂部尿道亦称舟状窝,同样来源于贯穿龟头的尿道板,但其上皮为复层鳞状上皮。
流行病学
AUV 与后尿道瓣膜(PUV)的发病率之比为 1:8;PUV 在每 8,000 至 25,000 例活产男婴中出现 1 例。前尿道憩室不常见,但在婴儿和儿童中,它是仅次于 PUV 的第二常见先天性尿道梗阻形式。到 1993 年为止,文献中报道的巨尿道病例不足 50 例,其中以舟状型更为常见。库珀氏腺导管囊肿和先天性尿道息肉都非常罕见。
前尿道瓣膜
前尿道瓣膜是前尿道最常见的先天性梗阻性病变,但其发生率比PUV低25-30倍。经典描述为:可表现为带孔的膈样膜,或源自尿道腹侧壁的黏膜瓣叶。其胚胎学尚不明确,因为前尿道瓣膜偶见位于憩室的远端。然而,受累前尿道段的尿道海绵体发育不全提示其处于尿道下裂谱系中的某一位置,或提示尿道黏膜与舟状窝上皮之间的结合不良。亦有提出扩张的球尿道腺破裂为其病因的观点。此类瓣膜可位于前尿道的任何部位:40%位于球部尿道,30%位于阴茎阴囊交界处,30%位于阴茎尿道;舟状窝受累者罕见。临床表现通常为梗阻性症状。患者常见尿滴沥、排尿困难、尿失禁、尿等待或尿潴留、尿流细弱,以及反复泌尿道感染;较大儿童还可出现遗尿、排尿后滴沥或生长发育不良。上尿路的继发性改变较为少见。然而,在新生儿期,梗阻可导致巨膀胱、膀胱破裂、重度输尿管肾积水、急性肾损伤或尿性腹水。排尿性膀胱尿道造影(MCUG)、逆行尿道造影和内镜检查是有用的诊断工具。在MCUG上,瓣膜本身可表现为沿腹侧壁的线样充盈缺损,但也可能仅表现为尿道管径的突然改变。
约三分之一的患者可见膀胱输尿管返流,且有一半患者可见上尿路扩张。内镜下,瓣膜呈现为薄膜样的、位于腹侧的瓣叶或组织瓣,偶尔被描述为虹膜样的膜性结构。必须仔细检查任何尿道憩室远端的尿道,因为冲洗液的逆向回流可能会将瓣膜结构压贴在尿道壁上。在膀胱充盈的情况下施以耻骨上按压,并打开内镜上的冲洗通道,可能更容易显示出该瓣膜结构。
此外,通过抬起或使用内镜电切环勾住该瓣膜,对于识别病变极为有用。经尿道切开瓣膜对大多数患者已足够,但必须注意切开要彻底,以避免遗留任何远端造成阻塞的唇缘。内镜治疗在多数情况下会使患者遗留尿道憩室。个别伴有巨大憩室且尿道海绵体缺损的患者,可能从开放修复中获益,该术式可同时重建瓣膜、憩室及其周围包绕组织。对于早产儿或体型较小的婴儿,可能需要行膀胱造口术,以便在婴儿能够容纳膀胱镜或接受进一步重建之前缓解梗阻。多达80%的前尿道瓣膜患儿在尿动力学检查中可见膀胱功能障碍、不稳定、反射亢进,以及顺应性与容量降低。由于临床表现较轻且更隐匿,AUV较PUVs总体肾功能保留率更高,约78%的患者在治疗后肾功能正常。
尿道憩室
较常见的为宽口型,通常位于阴茎-阴囊交界区,随着憩室逐渐扩张,其远端囊口边缘可导致瓣膜样梗阻。临床表现为梗阻症状或排尿后滴沥。较罕见的囊袋样病变具有狭窄的颈部,可发生于阴茎尿道全程的任何部位,包括舟状窝。临床表现多为尿路感染;少见与憩室内结石形成相关。治疗可经内镜切开或切除造成梗阻的瓣缘;少数情况下可经会阴入路行开放手术以切除大型憩室。
巨尿道
它是一种罕见的先天性畸形,其特征是在无明确梗阻的情况下阴茎尿道异常扩张。它可能与尿道海绵体缺如,或阴茎海绵体完全缺如有关。在这种情况下,阴茎几乎仅为一个松软的囊,外层为皮肤,内层为尿道黏膜。其与尿道憩室有部分相似之处,但对尿道的累及更为广泛且均一。将其分为梭形型与舟状型的分类反映了缺陷的严重程度及其对尿道海绵体和阴茎海绵体的影响。舟状型源于腹侧尿道海绵体的缺乏或缺如。梭形巨尿道还累及背侧尿道海绵体及阴茎海绵体。Adamson 和 Burge 描述了第三型,其中所有海绵体结构均完整。巨尿道的病因仍存一定争议。Stephens 提出,远端上皮索的管腔化延迟可能导致梗阻和近端扩张。另有观点认为,包绕尿道褶的中胚层发生胚胎学性停滞,会影响海绵体与勃起组织的发育。
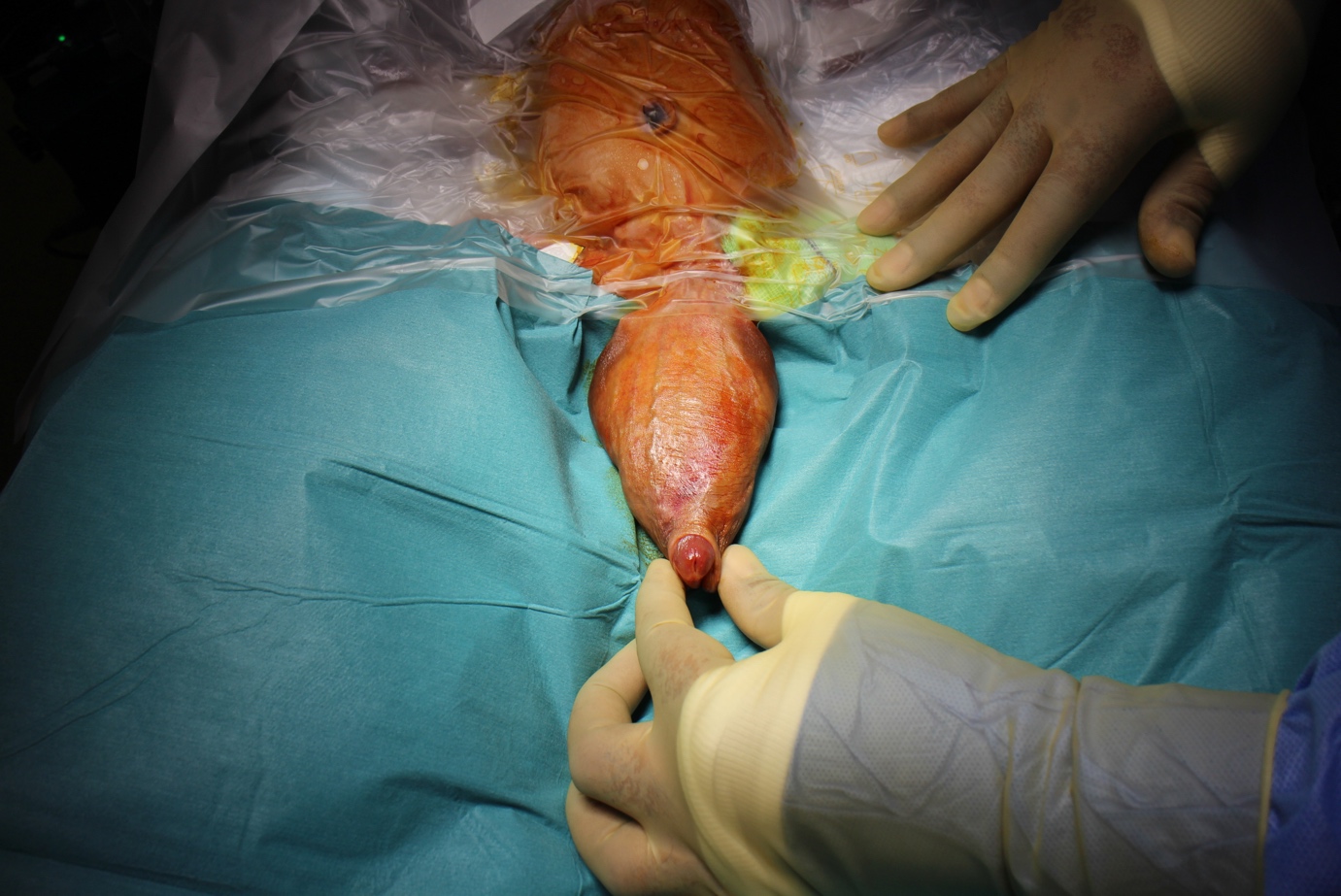
图 1 麻醉下检查显示累及阴茎干的显著巨尿道。
巨尿道(megalourethra)常与其他泌尿—直肠畸形相关,如肛门闭锁、梅干腹综合征和尿道瓣膜,并可伴有不同程度的梗阻性尿路病变。诊断多在体格检查时即被怀疑,尤其在观察患儿排尿时。尿道外口的位置可能正常,但仍存在病理性改变。柔软、细长的阴茎常提示罕见且更为严重的梭形型,其可触及阴茎海绵体明显不足。排尿时阴茎腹侧如气球样膨出是舟状型巨尿道的典型表现。为明确诊断并评估上尿路,应行排尿性膀胱尿道造影和肾脏超声检查。治疗的核心是按照尿道下裂修复的标准原则重建尿道与尿道海绵体。舟状型尿道可行纵向切开,并利用较好的背侧与侧方组织予以管状化。梭形型则因阴茎海绵体组织量不同而更具挑战性;从功能角度看,部分病例可能无法完全修复,成年后可通过植入阴茎假体进行阴茎海绵体重建而获益。
尿道球腺导管囊肿
Syringocoele(来自“syrongos”,意为管道,和“coele”,意为肿胀)是库珀腺导管远端部分的一种罕见囊性扩张。该腺体以外科医生兼解剖学家威廉·库珀(William Cowper)的名字命名,他在17世纪首次描述了它。库珀腺与女性的巴氏腺同源。这些外分泌腺是成对的尿道球腺,位于膜部尿道的两侧。主腺位于泌尿生殖膈上,副腺深位于海绵状球部尿道内。这些腺体的导管穿过外括约肌进入球部尿道,并在更远端1至2厘米(cm)处分别开口,或合并为单一导管开口。这些腺体在性兴奋时产生黏液样分泌物,构成射精前液的一部分,有助于润滑尿道以利精子通过。
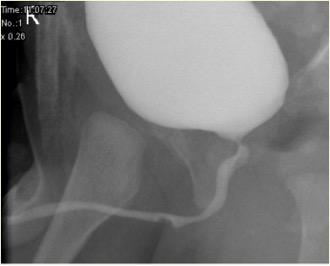
图 2 未破裂的尿道球腺管囊肿在 MCUG 中的表现
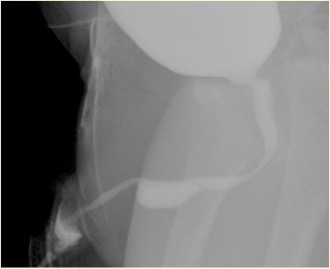
图 3 MCUG中破裂的库珀腺导管囊肿表现
库珀腺导管囊肿的真实发病率尚不清楚,这可能与该病罕见且对其不熟悉有关,并且还受到其他替代诊断存在的影响,如假道、尿道憩室或前尿道瓣膜;此外,在部分患者中还可能出现自发性消退。导管开口的梗阻可为先天性或获得性。导管上皮的增生可导致先天性潴留性囊肿以及狭窄近端的导管扩张。
感染或反复的尿道置管是获得性梗阻的原因。球尿道腺导管囊肿较为罕见,可表现为反复尿路感染、血尿、梗阻症状,或排尿后滴沥。少见情况下可为偶然发现。生命早期由球尿道腺导管囊肿导致的显著膀胱出口梗阻可引起慢性肾脏病和膀胱功能障碍。因此,对所有患者而言,早期诊断和及时治疗至关重要。一种实用的分类法考虑了临床表现和患者管理两个方面。分组为:梗阻型、非梗阻症状型和非梗阻无症状型。梗阻型包括所有出现膀胱出口梗阻体征或症状,或具有影像学或内镜学证据的患者。通常于婴儿早期出现。
相关异常包括膀胱壁增厚、多囊性发育不良肾(MCDK)、发育不良肾以及肾盂输尿管连接部(PUJ)梗阻。排尿性膀胱尿道造影(MCUG)是一项诊断性检查,可显示尿道”充盈缺损”的外观(未破裂的导管囊肿),或可见造影剂充盈破裂的囊肿并伴膀胱小梁形成、VUR 和排空不全。膀胱镜检查可予以证实。内镜治疗可采用多种技术处理导管囊肿,即 Bugbee 电极、剪刀、带钩尿道导管、导尿以及钬:YAG 激光。在内镜治疗过程中,将未破裂的导管囊肿切开开放,并可切开所有破裂导管囊肿的囊口唇缘。导管囊肿在婴儿中并非可忽视的情况,所有患儿必须得到恰当治疗并密切随访。在小儿泌尿外科实践中,评估前尿道梗阻时必须考虑导管囊肿。需对导管囊肿患儿进行密切随访,因为多达40%存在肾功能受损,其中近五分之二进展为慢性肾衰竭。少数存在膀胱功能障碍。
尿道闭锁与缺如
在胎内诊断出的肾脏异常和双侧肾积水的鉴别诊断中必须纳入尿道闭锁和尿道缺如。遗憾的是,除非存在其他使尿液能够从膀胱外流的通道,例如脐尿管未闭或尿直肠交通,否则这些病变与肾脏发育不相容。治疗将取决于具体畸形以及通过替代性尿路引流所能挽救的肾功能量。已认识到女性中尿道闭锁与梅干腹综合征之间的相关性,但很少有报道讨论这些病情危重婴儿的治疗结局。由于新生儿期的严重肾损害,这些儿童中的大多数最终需要肾移植。在男性中,渐进性导管扩张已被合理应用,且在女性中可能同样有效(PADUA—通过扩张尿道前部进行渐进性扩大)。该方法能否提供长期解决方案仍不清楚。
尿道重复畸形
这是一组罕见的尿道畸形。鉴于其解剖变异多样,目前没有共同的胚胎学机制能够解释所有类型的尿道重复。Johnson 提出,生殖嵴与尿道褶融合不良可导致形成两个独立通道;Lowsley 则认为,在生殖嵴内折过程中泌尿生殖板持续存在可作为解释。尿道重复可与阴茎或膀胱的完全重复同时出现,属于更为严重的异常。尿道重复分为两型:矢状型和并列型。大多数重复发生于矢状面,且仅有单一阴茎。在这种一上一下的情形中,优势尿道开口位于尿道下裂位置,而副尿道开口位于解剖学正位。许多非优势的背侧尿道在距膀胱近端即盲端终止。但如果其确实与膀胱相通,患儿常因该副通道而出现尿失禁。在并列型中,重复尿道并行走行。膀胱和尿道的完全重复较为罕见,女性中尤为少见。
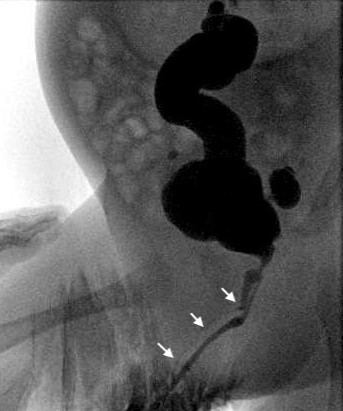
图 4 膀胱造影检查显示Y型尿道重复,箭头指向(背侧)非优势尿道。
在某些病例中,两条尿道从膀胱分别起始,且在全程彼此分离;而在另一些病例中,重复的尿道在远端汇合形成单一通道。在所谓的'纺锤形'类型中,尿道先分为两个组成部分,随后在更远端再次汇合;而在'Y'型重复中,副尿道从主通道分出,开口于肛周或会阴部。在伴有上裂性尿道口的病例中,可能发现耻骨联合增宽,提示与膀胱外翻—尿道上裂复合体有关。手术重建取决于个体解剖,但几乎总是需要切除较狭窄的副尿道。在重建时偶尔需要进行内镜检查以全面了解病变。若仅因排尿时尿线轻度分叉而发现问题,可能无需修复。此类考虑通常仅适用于两处尿道外口彼此紧邻的患者;即便如此,这些患者也可能从简单的尿道口成形术中获益,将两个开口合并为一个。随着两个开口分离程度的增加,修复的复杂性也随之提升,尤其当主导的腹侧尿道外口非常近端时。在这种情况下,通常需要将腹侧会阴尿道从直肠旁游离并移至更正位的位置,并采用多种尿道成形术技术重建其余尿道。若包皮完整,则十分有用。若发现该畸形,患者不应在新生儿期接受包皮环切术。对于既往已行包皮环切或手术的部分患者,可能需要黏膜移植。对于包皮皮肤长度不足的部分患者,也可能需要黏膜移植。
为了重建腹侧尿道所需的较为广泛的分离,通常可获得良好的显露,以便切除背侧副尿道。该结构也可在近端与腹侧尿道行吻合。Passerine-Glazel 及其同事描述了对背侧尿道进行逐步序贯扩张,使其可用于排尿。我们在大多数情况下更倾向于游离并重建优势尿道。
尿道外口狭窄
尿道外口狭窄最常见于接受包皮环切的男孩,由尿布性皮炎引起。首发症状通常为尿流减弱或尿线偏斜。尿道外口切开术和尿道外口成形术是可靠的治疗方法。
前尿道狭窄
前尿道狭窄多为特发性(或许为先天性)或外伤性所致。然而,尿道下裂修复术后的狭窄在前尿道也较为常见;其原因可能是修复术本身,或作为尿道支架使用的导尿管所致。前尿道狭窄通常引起刺激症状(血尿、排尿痛、尿湿〔尿失禁〕)或梗阻症状(排尿用力、尿潴留)。通过尿道的放射学成像或内镜检查最易确诊。尿流率测定可能显示梗阻性流型,但即便存在狭窄,流率也可能正常;因此,不能依赖尿流率测定来排除狭窄。直视下尿道内切开术仅对约一半病例有效,但若仅实施一次,可能并无害处。扩张治疗似乎并非恰当的处理方式,因为需要多次反复进行。切除狭窄段后的端端尿道吻合术在解剖条件允许时是最有效的治疗,即便是尿道下裂术后狭窄亦然。当无法实施尿道吻合时,采用口腔颊黏膜或皮肤的补片移植通常是成功的。尿道修补中最好避免使用PDS作为缝线材料。
尿道息肉
尿道息肉是男性尿道的另一种不常见异常。它们可能为先天性,但也在成人中有报道,提示其可以为后天获得性,或缓慢生长直至大到足以引起症状。这些纤维-内皮性病变常起源于精阜,通常为纤维肌性核心,其表面覆以移行上皮。小的息肉通常在为其他原因进行的内镜检查中偶然发现。相反,范围更大的病变,其息肉样头部连于一根延长的蒂上自由漂浮,往往会突出穿过膀胱颈,导致急性、短暂的尿潴留发作。其他症状包括排尿用力、尿急和血尿。亦可出现明显的尿道出血。诊断依靠MCU或膀胱尿道镜检查。排尿性膀胱尿道造影常显示尿道内的充盈缺损,位置可变。诊断可通过膀胱镜检查予以证实。大多数息肉可经内镜切除(经尿道切除),但对于较大的息肉,可能需要开放式经膀胱入路。
要点
- 前尿道瓣是前尿道最常见的先天性梗阻性病变,通常表现为梗阻症状。对大多数患者,经尿道瓣切开术已足够。
- 尿道憩室可表现为更常见的宽口型,或囊状型。治疗为内镜下切开或切除阻塞性瓣样缘,少数情况下需采用会阴开放入路切除大型憩室。
- 巨尿道是一种罕见的先天性异常,其特征是阴茎尿道在无结构性梗阻的情况下出现异常扩张。为确立诊断并评估上尿路,需行排尿性膀胱尿道造影和肾脏超声检查。治疗重点是依据尿道下裂修复的标准原则重建尿道和尿道海绵体。
- 库珀氏腺导管囊肿是一种发生于库珀氏腺导管远端的罕见囊性扩张。对所有患者而言,早期诊断与及时治疗至关重要。内镜治疗可采用多种技术处理此类囊肿,即 Bugbee 电极、剪刀、带钩尿道导管、导尿,以及钬:YAG 激光。
- 尿道闭锁与发育缺如应纳入胎儿期(宫内)诊断出的肾脏异常和双侧肾积水的鉴别诊断。处理取决于具体畸形以及通过替代性尿路引流所能挽救的肾功能量。在男性中,逐步导管扩张已被合理应用,对女性也可能同样有效(PADUA-通过扩张前尿道进行渐进性增大)。
- 尿道重复是一组罕见的尿道畸形。随着两个外口分离程度加大,修复的复杂性也随之增加,尤其当优势的腹侧尿道口非常近端时。
- 尿道口狭窄最常见于已行包皮环切的男孩,多由尿布性皮炎所致。尿道口切开术和尿道口成形术是可靠的处理方法。
- 前尿道狭窄多为特发性(或许为先天性)或由外伤所致。切除狭窄段后行端端尿道吻合术是在解剖上可行时最有效的治疗,即使对于尿道下裂术后狭窄亦然。
- 尿道息肉是男性尿道的不常见畸形。这些纤维-内皮性病变常起源于精阜。诊断依靠 MCU 或膀胱尿道镜检查。大多数息肉可经内镜切除(经尿道切除),但对于较大的息肉,可能需要经膀胱开放入路。
参考文献
- Antón-Juanilla M, Lozano-Ortega JL, Galbarriatu-Gutiérrez A. A Urresola-Olabarrieta, Anterior urethral trauma in childhood: presentation of two cases. Cir Pediatr 2020; 1;33(4):200-203.
- Ansari MS, Yadav P, Srivastava A, Kapoor R, Shekar PA. Etiology, and characteristics of pediatric urethral strictures in a developing country in the 21st century. J Pediatr Urol 2019; 15: 403.e1–403.e8. DOI: 10.1016/j.jpurol.2019.05.020.
- Vetterlein MW, Weisbach L, Reichardt S, Fisch M. Anterior Urethral Strictures in Children: Disease Etiology and Comparative Effectiveness of Endoscopic Treatment vs Open Surgical Reconstruction. Pediatr 2019. DOI: 10.3389/fped.2019.00005.
- Kaplan GW, Brock JW, Fisch M, Koraitim MM, Snyder HM. SIU/ICUD Consultation on Urethral Strictures: Urethral Strictures in Children. Urology 2014; 83. DOI: 10.1016/j.urology.2013.09.010.
- Perlman S, Borovitz Y, Ben-Meir D, Hazan Y, Bardin RNR, Brusilov M, et al.. Prenatal diagnosis and postnatal outcome of anterior urethral anomalies. 2020; 40 (2): 191–196. DOI: 10.1002/pd.5582.
- Thomas D, Duffy P, Rickwood A. Essentials of Paediatric Urology. 2008: 117–120. DOI: 10.1046/j.1464-410x.2002.t01-1-03073_1.x.
- Gillenwater J, Howards S, Grayhack J, Mitchell M. Adult and Pediatric Urology ". 4th ed., USA: Lippincott Williams & Wilkins Publishers; 2002, DOI: 10.1001/jama.1991.03460240115044.
- Stringer M, Oldham K, Mouriquand P. Pediatric Surgery and Urology: long – term outcomes". 2nd ed., USA: Cambridge University Press; 2006, DOI: 10.1016/s0002-9610(99)00082-3.
- Berrocal T, López-Pereira P, Arjonilla A, Gutiérrez J. Anomalies of the distal ureter, bladder, and urethra in children: embryologic, radiologic and pathologic features". Radiographics 2002. DOI: 10.1148/radiographics.22.5.g02se101139.
- Alonso AR, Outeda EC, Blanco AG, Martín CB, Franco JL, Pérez MAC, et al.. Duplicidad de uretra masculina. Actas Urológicas Españolas 2002; 26 (1): 69–73.
最近更新时间: 2025-09-22 08:00