
27: Síndrome de Prune Belly
Este capítulo durará aproximadamente 30 minutos para leer.
Introducción
El síndrome del abdomen en ciruela pasa (PBS), también denominado síndrome de Eagle-Barrett, es una afección multisistémica poco frecuente, típicamente caracterizada por una constelación de anomalías con grados variables de gravedad; los hallazgos principales son la deficiencia de la musculatura abdominal, testículos intraabdominales bilaterales y un tracto urinario anómalo.1,2 Otras anomalías asociadas no genitourinarias afectan el aparato respiratorio, el tracto gastrointestinal, el sistema cardíaco y el sistema musculoesquelético.3 La gravedad de la enfermedad existe a lo largo de un amplio continuo, con algunos niños que no sobreviven al período neonatal y otros que se ven mínimamente afectados.4
Embriología
Se han propuesto varias teorías predominantes sobre la embriogénesis del PBS aunque ninguna de ellas tiene aceptación universal, y existe cierta superposición entre ellas. Las cuatro teorías principales son 1, una obstrucción uretral posterior temprana in utero (probablemente una próstata hipoplásica/displásica o una uretra anormal), que da lugar a dilatación vesical proximal, ureteral y renal con desarrollo deficiente asociado de la pared abdominal 2, un defecto primario en el mesodermo de la placa lateral, que es el precursor de los uréteres, la vejiga, la próstata, la uretra y el gubernáculo 3, un defecto intrínseco del tracto urinario que conduce a dilatación ureteral y ascitis fetal y 4, un defecto del saco vitelino.5,6
Epidemiología
La incidencia de la PBS se ha reportado en 1 de cada 29.000 a 1 de cada 40.000 nacidos vivos, similar a la de la extrofia vesical, con el 95% de los pacientes afectados siendo varones.1,2,7 Las mujeres representan menos del 5% de todos los casos de PBS y presentan deficiencia de la pared abdominal y un tracto urinario dilatado y dismórfico sin anomalías gonadales asociadas.8 Se observa una mayor incidencia en gemelos, en personas negras y en niños nacidos de madres más jóvenes.9 Aunque la PBS a menudo se presenta como un evento esporádico, también se sospecha una contribución genética con mutaciones en genes que codifican factores de transcripción del músculo liso, filamentos contráctiles y enzimas/receptores neuronales.10,11
Características clínicas y patogénesis
Anomalías genitourinarias
Riñones
El espectro de anomalías renales en pacientes con PBS abarca desde un parénquima renal normal hasta displasia. Alrededor de la mitad de los casos presentan displasia renal, que suele corresponder a las variedades tipo II y IV de Potter, y esto también puede variar entre ambos lados.1,2 Mientras que la variedad tipo II, con pocos nefrones y desorganización parenquimatosa, es más indicativa de un defecto mesenquimatoso renal, la variedad tipo IV, con quistes corticales y tubulares, se asocia a obstrucción de la salida, donde no ha habido descompresión uracal.12 Aunque el sistema colector renal está característicamente dilatado, a menudo de forma marcada, el grado de dilatación no se correlaciona con el grado de displasia renal y la morfología calicial puede estar bien preservada, incluso en presencia de dilatación masiva. Aunque la hidronefrosis suele ser no obstructiva, también puede presentarse obstrucción primaria o secundaria de la unión pieloureteral. Sin embargo, es importante señalar que es la infección renal, más que la obstrucción, la que representa el mayor riesgo para la función renal en estos pacientes.1,2
Uréteres
Los uréteres suelen estar dilatados, tortuosos y redundantes. Aunque la dilatación masiva y la estenosis pueden ocurrir en todos los niveles, las porciones proximales (superiores) de los uréteres suelen ser menos anómalas que los segmentos distales. El reflujo vesicoureteral (VUR) está presente en el 75% de los niños con PBS.12 La obstrucción no es común, pero se ha reportado en las uniones ureteropélvica y ureterovesical. Los cortes histológicos demuestran escasez de células de músculo liso y un aumento de la proporción de colágeno respecto al músculo liso en los segmentos distales más afectados, especialmente en los que presentan reflujo, con células de músculo liso de aspecto más normal en los segmentos proximales.13 Este hecho es crítico cuando se emprende la reconstrucción ureteral. Se considera que la disminución del número de miofibrillas gruesas y finas observada en el examen ultraestructural contribuye a la peristalsis ineficaz debido a la deficiente coaptación de la pared ureteral, lo que resulta en estasis de la vía urinaria superior y puede conducir a infección. Además, en muchos pacientes la gravedad de las anomalías del tracto urinario no es proporcional al grado de flacidez de la pared abdominal.14
Vejiga
La vejiga suele verse muy aumentada de tamaño, con un seudodivertículo a nivel del uraco y un uraco permeable en el 25% al 30% de los pacientes.12 A diferencia de la imagen observada en vejigas obstruidas por otras causas, pese a ser muy gruesa, el contorno vesical es liso. Durante la micción, el cuello vesical se abre ampliamente hacia una uretra prostática dilatada.1,2 Histológicamente, en ausencia de obstrucción, la vejiga presenta una relación aumentada de colágeno respecto de las fibras musculares; sin embargo, puede observarse hipertrofia del músculo liso cuando existe obstrucción. En la cistoscopia, el trígono está ensanchado, con los orificios ureterales desplazados superior y lateralmente, lo que posiblemente contribuye a la alta incidencia de reflujo vesicoureteral. Los hallazgos característicos en la evaluación urodinámica incluyen una vejiga de complacencia normal con primera sensación de micción retrasada y gran capacidad.15 La eficiencia miccional de estas vejigas es variable; algunas se vacían por completo, mientras que otras presentan un residuo posmiccional significativo, posiblemente debido a una obstrucción relativa de la salida y a la incapacidad del músculo detrusor para generar presión suficiente durante la micción en estos pacientes.16 El término “micción desequilibrada” se usa habitualmente para referirse a la primera situación en la que una resistencia relativa a la salida impide un vaciado vesical eficaz.1,2 Aunque el 50% de los pacientes con síndrome de Prune-Belly miccionan espontáneamente con presiones miccionales y tasas de flujo normales y bajos residuos posmiccionales, se ha observado que el deterioro de la micción equilibrada puede ocurrir con el crecimiento, resultando en residuos posmiccionales significativos, lo que enfatiza la necesidad de una evaluación periódica.16
Próstata y órganos sexuales accesorios
La dilatación uretral posterior se debe a hipoplasia prostática, probablemente relacionada con un desarrollo mesenquimal-epitelial anormal.12,13,14,15,16,17,18,19 Los cortes histológicos revelan pocos elementos celulares prostáticos, con una disminución de las células epiteliales y del músculo liso y un aumento de las células del tejido conectivo.19 Se ha observado que aproximadamente el 20% de los pacientes también presentan lesiones obstructivas en la uretra posterior distal en forma de atresia uretral, válvulas, estenosis uretral, membrana uretral y divertículo uretral. La falta de tejido parenquimatoso prostático puede provocar una angulación de la uretra durante la micción, lo que Stephens denominó válvula tipo IV.12 Se considera también que la hipoplasia prostática y la incompetencia del cuello vesical asociada son un factor en el fracaso eyaculatorio/ eyaculación retrógrada en pacientes con PBS.1,2 Los órganos sexuales accesorios, como el conducto deferente y las vesículas seminales, suelen estar atrésicos, aunque cualquiera de ellos puede estar dilatado o engrosado. El epidídimo puede estar mal adherido al testículo y también puede haber falta de continuidad entre los conductillos eferentes y la red testicular.20
Uretra anterior
Aunque la uretra anterior del niño con PBS suele ser normal, se han descrito varias anomalías del segmento uretral; las más comunes son la atresia o hipoplasia uretral y la megalouretra.21,22,23,24,25 Se ha postulado que la atresia o hipoplasia uretral ocurre porque la uretra no se utiliza, más que por estar malformada. La atresia uretral suele ser letal si no se asocia a descompresión en forma de un uraco permeable o, a veces, incluso a rotura vesical espontánea con formación de fístula.22
Una característica única en el PBS es la aparición de megalouretra.26 Se ha propuesto que una obstrucción transitoria in útero de la unión entre la uretra glandular y la peneana provoque megalouretra. Se observan dos variantes de megalouretra en pacientes con PBS, a saber, la fusiforme (Figura 1) y la escafoide (Figura 2), (Figura 3) variante.24 El tipo fusiforme es una deficiencia del cuerpo cavernoso, así como del cuerpo esponjoso, probablemente resultante de una deficiencia mesenquimal de los pliegues uretrales, y clínicamente se observa como dilatación de todo el falo durante la micción. Por otro lado, la variante escafoide, que probablemente surge de una deficiencia mesenquimal de los tejidos de sostén uretrales que conduce a deficiencia del cuerpo esponjoso únicamente con preservación del glande y de los cuerpos cavernosos, se caracteriza por dilatación de la uretra ventral durante la micción.26
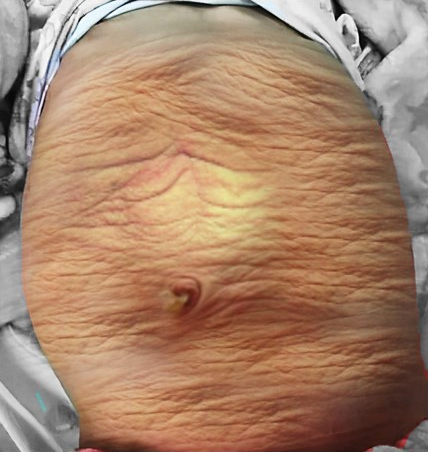
Figura 1 Fotografía clínica de un niño con síndrome del abdomen en ciruela pasa
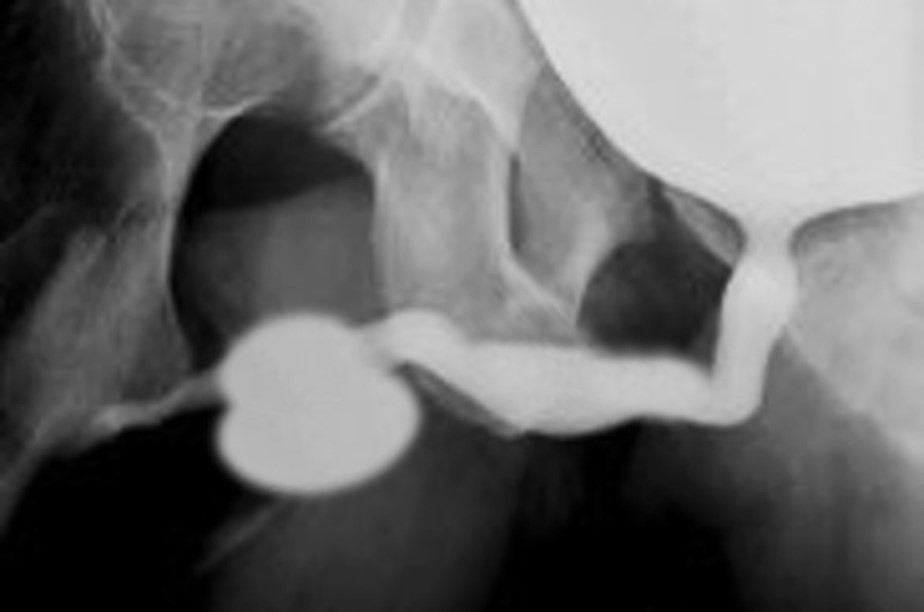
Figura 2 Megalouretra fusiforme
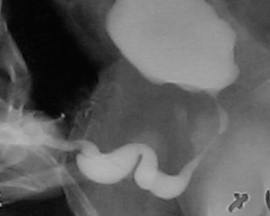
Figura 3 Megalouretra escafoidea
Testículos
Los testículos intraabdominales bilaterales situados sobre los vasos ilíacos y adyacentes a los uréteres dilatados se consideran uno de los hallazgos típicos en el PBS.1,2 Aunque desde hace tiempo se ha propuesto que fuerzas mecánicas, como una obstrucción mecánica debida a megacistis y una pobre presión intraabdominal secundaria a anomalías de la pared abdominal, son la etiología de este fallo del descenso testicular, el hecho de que algunos pacientes con las anomalías típicas del tracto urinario y de la musculatura abdominal presenten testículos descendidos genera cierta duda sobre factores puramente mecánicos.1,2 En cuanto a si los testículos en niños con PBS son intrínsecamente diferentes de los de niños sin PBS, Pak et al compararon la histología de testículos intraabdominales y no encontraron diferencias en el recuento de células germinales, espermatogonias y células de Leydig entre los testículos con PBS y los testículos fetales intraabdominales sin PBS.27,28 Sin embargo, Orvis et al observaron disminución del número de espermatogonias e hiperplasia de células de Leydig en testículos fetales con PBS, lo que sugiere una anomalía testicular intrínseca.29 Con respecto al riesgo de malignidad en estos testículos, aunque el riesgo puede ser relativamente bajo debido a la ausencia de epitelio germinal, es necesaria una orquidopexia temprana y un seguimiento a largo plazo para reducir el riesgo de malignidad testicular y mejorar su detección.30,31
Defecto de la pared abdominal
El aspecto de la pared abdominal es una característica distintiva en los niños con PBS. Con mayor frecuencia, la deficiencia muscular es desigual y parcheada, con los segmentos musculares mediales e inferiores típicamente más deficientes, y puede ser desproporcionada con respecto a las anomalías del tracto urinario. Las áreas más gravemente afectadas pueden presentar piel, grasa subcutánea y una única capa fibrosa sobre el peritoneo, y el examen histológico mediante microscopía electrónica demuestra un patrón inespecífico de desarreglo de miofilamentos, desorganización de la línea Z y proliferación mitocondrial.32,33 A pesar de estos problemas de la pared abdominal, en estos niños se observa una buena cicatrización sin tendencia a infecciones ni a hernias incisionales. El aspecto típico al nacer es el de piel arrugada y redundante, con un abdomen que se abomba en los flancos. A medida que los pacientes crecen, algunos muestran una mejoría del tono y del aspecto abdominal, con menos arrugas, mientras que en otros el abdomen adquiere más un aspecto de abdomen globuloso.1,2 La marcha por lo general no se ve afectada, aunque puede estar retrasada, y los niños tienden a girar hacia un costado y a usar los brazos para incorporarse desde la posición supina. El escaso sostén de la pared torácica inferior puede provocar ensanchamiento del reborde costal, y la consiguiente disminución de la eficacia de la tos hace que estos niños sean más vulnerables a las enfermedades respiratorias.
Anomalías extragenitourinarias
De todos los niños con PBS, el 75% presenta anomalías extraurinarias.1,2,4 Tras el evidente defecto de la pared abdominal, los problemas más frecuentes son cardíacos, pulmonares y ortopédicos (Tabla 1).34,35,36,37,38,39,40 Además de estas morbilidades específicas de órgano, casi el 50% de los niños con PBS nacen prematuros, lo que contribuye de forma significativa a las comorbilidades.
Tabla 1 Anomalías extragenitourinarias comunes observadas en el síndrome de abdomen en ciruela pasa
| Sistema | Defectos comunes | Observaciones |
|---|---|---|
| Anomalías cardíacas (10%) | Persistencia del conducto arterioso, defectos septales auricular y ventricular, defecto del tabique ventricular y tetralogía de Fallot | Las anomalías cardíacas al nacimiento pueden tener prioridad sobre los problemas urológicos. |
| Anomalías pulmonares (55%) | Hipoplasia pulmonar. Neumotórax y neumomediastino pueden observarse con o sin hipoplasia pulmonar | La hipoplasia pulmonar puede resultar de oligohidramnios grave relacionado con displasia renal o con obstrucción grave de la salida vesical y puede causar muerte neonatal. En casi la mitad de los recién nacidos con PBS, se requerirán intubación y soporte ventilatorio mecánico, con las morbilidades asociadas. La incapacidad para generar una presión intraabdominal significativa y las anomalías musculoesqueléticas asociadas, como escoliosis y anomalías de la caja torácica, con mayor frecuencia pectus excavatum, pueden contribuir a un mayor riesgo de neumonía y atelectasia lobar en estos niños. |
| Anomalías gastrointestinales (30%) | Las anomalías resultan de la rotación incompleta del intestino medio que da lugar a un mesenterio amplio, lo que produce mayor movilidad intestinal con malrotación intestinal, vólvulos, atresias y estenosis. También se ha informado torsión esplénica relacionada con una fijación mesentérica anormal. Se han reportado onfalocele, gastrosquisis, situs inversus abdominis, VACTERL (defectos vertebrales, atresia anal, defectos cardíacos, fístula traqueoesofágica, anomalías renales y anomalías de las extremidades) y anomalías anorrectales. | Con una capacidad limitada para generar presión intraabdominal como resultado de la hipoplasia de la musculatura abdominal, el estreñimiento se convierte en un problema de por vida y puede conducir a megacolon adquirido. |
| Anomalías ortopédicas (65%) | Los hoyuelos en la cara lateral de las rodillas son un hallazgo común en el oligohidramnios y se observan con frecuencia en pacientes con PBS junto con otros defectos como el pie equinovaro (26%), displasia de cadera (5%) y escoliosis congénita (4%). Hipoplasia, ausencia o amputación de las extremidades inferiores. | Dado que la mayoría de estos defectos musculoesqueléticos son unilaterales, la causa más probable de estos defectos es el efecto compresivo del oligohidramnios. Otro mecanismo propuesto para las anomalías de las extremidades inferiores ha sido la compresión de los vasos ilíacos externos por la vejiga distendida, comprometiendo así el suministro sanguíneo a las extremidades inferiores. Además, algunas de estas malformaciones, como la osteodistrofia renal, la luxación de la cadera, la escoliosis y el pectus excavatum o carinatum, tienden a manifestarse o empeorar con el crecimiento. |
Diagnóstico y evaluación
Prenatal
Aunque el diagnóstico de PBS mediante ecografía se ha reportado tan temprano como a las 11-12 semanas de gestación, la precisión diagnóstica varía del 30% al 85%, ya que el PBS se presenta prenatalmente con muchos hallazgos ecográficos comparables a los de la obstrucción del tracto urinario inferior (LUTO) debida a otras causas, incluidas las válvulas uretrales posteriores, el ureterocele y la atresia uretral.40,41,42,43,44 Se debe considerar el diagnóstico prenatal de PBS siempre que se identifiquen claramente las siguientes anomalías ecográficas: oligohidramnios, anomalías urinarias (dilatación del tracto urinario, megacistis, hidroureteronefrosis bilateral) y ausencia de musculatura abdominal. El diagnóstico precoz no solo mejora la supervivencia al motivar al clínico tratante a realizar el parto en un centro terciario para un manejo multidisciplinario oportuno de los recién nacidos afectados, sino que también brinda a los futuros padres la opción de interrupción voluntaria del embarazo, si así lo desean.43
Recién nacido
El aspecto de la pared abdominal es diagnóstico en el recién nacido, independientemente de si el diagnóstico era conocido prenatalmente. Los niños afectados con PBS representan un desafío único y requieren atención multidisciplinaria inmediata por parte de un equipo que incluya a un neonatólogo, urólogo, nefrólogo, así como cardiólogo y ortopedista pediátricos cuando esté indicado. Dada la amplia gama de gravedad de la enfermedad, debe realizarse en el periodo neonatal una evaluación multisistémica para descartar malformaciones cardíacas, pulmonares u otras asociadas significativas. Es necesaria una radiografía de tórax inmediata para descartar neumotórax y neumomediastino asociados. Como ocurre con LUTO, la evolución posnatal inicial está determinada por la gravedad de las comorbilidades, como la hipoplasia pulmonar, con peores resultados en aquellos con prematuridad e hipoplasia pulmonar grave.45,46,47
El oligohidramnios prenatal en estos lactantes indica una función renal alterada. Las mediciones iniciales de creatinina reflejan la función renal materna y es necesario realizar muestreos repetidos. Si tras 48–72 horas la creatinina sérica es mayor de 1.0 en el recién nacido a término o 1.5 en el recién nacido pretérmino, existe algún grado de insuficiencia renal. Un ascenso progresivo de la creatinina sérica durante las primeras semanas de vida presagia un mal pronóstico. Si el nadir inicial de creatinina es inferior a 0.7 mg/dL, entonces es poco probable una insuficiencia renal posterior.48 Se envía orina para cultivo basal y se inicia profilaxis antibiótica. La medición de electrolitos séricos y urinarios al nacimiento puede ser útil para evaluar la conservación de sodio, lo que sugiere una función renal adecuada.
Evaluación diagnóstica
Una evaluación urológica exhaustiva se lleva a cabo tras la estabilización del paciente debido a otras comorbilidades. Inicialmente se requieren el examen físico y la ecografía del tracto urinario.46 Las ecografías renales proporcionan información sobre el grosor cortical, la presencia de cambios quísticos y el tamaño renal. Los exámenes ecográficos antes y después de la micción orientan sobre la presencia de reflujo vesicoureteral y residuo urinario posmiccional.
Inicialmente, se realiza una cistouretrografía miccional (VCUG) en aquellos lactantes con función renal anormal para asegurarse de que no esté presente una verdadera obstrucción anatómica uretral. Esta “variante letal” y la posible confusión con válvulas uretrales posteriores que “simulan” el síndrome de abdomen en ciruela pasa deben diagnosticarse, ya que el tratamiento cambia de forma drástica. Además, la VCUG demuestra reflujo vesicoureteral en hasta el 85% de los pacientes y evalúa el vaciamiento vesical. Todos los lactantes son tratados con antibióticos antes de la cateterización estéril para disminuir el riesgo de infección. En general, la evaluación radiológica del tracto urinario se adapta a la filosofía de manejo del cirujano urólogo tratante. Se evitan los estudios con contraste que requieren instrumentación riesgosa en aquellos lactantes con buena función renal.22
Después de la evaluación inicial, una gammagrafía renal con mercaptoacetiltriglicina (MAG3) puede aportar información tanto funcional como anatómica. Se pueden evaluar el flujo sanguíneo, la función renal diferencial y el drenaje en respuesta a furosemida (Lasix), y compararlos con estudios posteriores. Se han señalado las limitaciones de la renografía diurética para demostrar obstrucción en pacientes con síndrome de prune belly.49 Ocasionalmente, la prueba de Whitaker es necesaria como estudio definitivo para demostrar una obstrucción significativa, aunque esto es principalmente de importancia histórica. Se realiza una gammagrafía renal con ácido dimercaptosuccínico (DMSA) en lugar de una urografía excretora (IVP) para evaluar el tamaño renal, la masa tubular y la función diferencial. Los estudios comparativos son invaluables para evaluar el crecimiento y las cicatrices renales, ya que estos pacientes requieren seguimiento a largo plazo. Algunos grupos han abogado por la urografía por resonancia magnética (MRU) en estos pacientes debido a su alta resolución y a los excelentes datos funcionales y anatómicos que permiten una visualización detallada de las anomalías del tracto urinario superior, lo cual también puede ser útil en la planificación quirúrgica.50 Como se muestra en Tabla 1, existen tres categorías principales de presentación neonatal según lo descrito por Woodard.1,2
Manejo y resultados
Manejo prenatal
Intervención intrauterina
Con la mejora de las técnicas avanzadas de imagen y diagnóstico materno-fetales, el síndrome del abdomen en ciruela pasa puede sospecharse mediante estudios de imagen en etapas tempranas del embarazo. La intervención fetal suele abordar el oligohidramnios o anhidramnios asociados a hipoplasia pulmonar, la principal causa de mortalidad perinatal. Los criterios para la intervención intrauterina incluyen gestación en el segundo o tercer trimestre, oligohidramnios grave, megacistis, hidronefrosis avanzada, cariotipo normal, alcance limitado de otras anomalías congénitas detectadas y análisis seriados favorables de orina fetal aspirada.51,52,53 Aunque existen varios informes de procedimientos de derivación vesicoamniótica para uropatía obstructiva fetal, las revisiones no han logrado documentar un efecto beneficioso de la intervención fetal sobre la función renal posterior y tampoco puede asegurarse la función pulmonar a pesar de la restauración de niveles normales de líquido amniótico. Dado que la mayoría de los pacientes con síndrome del abdomen en ciruela pasa no presentan evidencia demostrable de obstrucción y la función renal no se correlaciona con el grado de dilatación presente en el tracto urinario, deben sopesarse cuidadosamente los riesgos y beneficios de la intervención in útero.53
Manejo posnatal
Los objetivos generales del manejo son preservar la función renal y prevenir la infección. El logro de estos objetivos es posible mediante una variedad de enfoques terapéuticos, que van desde la vigilancia expectante hasta la reconstrucción quirúrgica inmediata del tracto urinario. Se evita la intervención quirúrgica temprana en el recién nacido, a menos que se presente un aumento de la creatinina o una infección, lo que requiere una vesicostomía temprana. Como se muestra en Tabla 2, el manejo en estos pacientes depende de la categoría de gravedad al momento de la presentación.
Tabla 2 Presentación y manejo de distintas categorías basadas en los criterios de presentación neonatal de Woodward
| Categoría de gravedad | Presentación | Manejo | Seguimiento y desenlaces |
|---|---|---|---|
| I | Oligohidramnios marcado secundario a displasia renal y/o obstrucción de la vía de salida que resulta en hipoplasia pulmonar grave y anomalías esqueléticas | En esos pocos lactantes que sobreviven a la agresión pulmonar, se ha intentado derivación urinaria alta mediante pielostomía cutánea; sin embargo, la recuperación de la función renal no suele ser posible debido a la displasia renal subyacente grave. | Estos neonatos a menudo fallecen en pocos días y las intervenciones son limitadas. Asociados con oligohidramnios prenatal e hipoplasia pulmonar posnatal, suelen morir en el período posnatal inmediato por complicaciones pulmonares. |
| La facies de Potter puede estar presente y es secundaria al oligohidramnios. Las características incluyen micrognatia, ojos muy separados, fisuras palpebrales aplanadas, epicanto prominente, puente nasal aplanado, orejas de implantación baja con falta de cartílago y deformidades esqueléticas. | Solo puede ser necesaria la cateterización | ||
| Los casos de atresia uretral suelen pertenecer a esta categoría más grave. | |||
| II | Insuficiencia renal moderada e hidroureteronefrosis moderada-grave. La hipoplasia pulmonar no es un rasgo prominente. | Se indican antibióticos profilácticos a largo plazo y se vigilan seriadamente para detectar infección y cualquier disminución de la función renal. | Se requiere vigilancia posoperatoria de por vida para evaluar la función renal y el drenaje óptimo de las vías urinarias superior e inferior con estudios funcionales y anatómicos. |
| Se persigue una reconstrucción agresiva del tracto urinario cuando hay documentación de infección, falta de crecimiento renal adecuado o disminución de la función renal. | |||
| Sin embargo, la reconstrucción quirúrgica suele posponerse hasta que el lactante tenga 3 meses de edad o más para permitir la maduración pulmonar. | |||
| Si la insuficiencia renal es grave o hay infección inicial, se recomienda un período de derivación urinaria mediante vesicostomía cutánea. | |||
| Las ureterostomías cutáneas probablemente aportan poco para mejorar el drenaje. Además, el uréter superior es anatómica e histológicamente el más normal y debe preservarse si posteriormente se elige una reconstrucción a medida. | |||
| III | Rasgos leves de la tríada o formas incompletas | Constituyen la mayoría de los pacientes y demuestran buena función renal, a pesar de la dilatación marcada del tracto urinario superior e inferior. | Similar a la categoría 2, se requiere vigilancia de por vida. |
| La función renal suele ser normal o estar levemente afectada y no hay insuficiencia pulmonar. | La intervención quirúrgica temprana para reconstruir el tracto urinario no suele ser necesaria y estos pacientes se manejan con profilaxis antibiótica a largo plazo y evaluación seriada de la función renal mediante técnicas de imagen selectivas. | La aparición de infecciones urinarias o la disminución de la función renal obliga a realizar una evaluación específica adicional en forma de estudio urodinámico o renograma diurético para documentar obstrucción del tracto inferior o superior que pueda requerir intervención. | |
| Estos pacientes se benefician de la orquidopexia y la abdominoplastia tempranas, ya que la corrección de la pared abdominal puede mejorar la defecación y la eficacia de la micción. |
Intervenciones quirúrgicas en el síndrome de abdomen en ciruela pasa
Tracto urinario
Un hallazgo característico en el PBS es la dilatación de todo el tracto urinario a baja presión, que se extiende desde la pelvis renal proximalmente hasta la uretra distalmente. La vejiga suele estar aumentada de tamaño e hipotónica, con complacencia elevada y reflujo vesicoureteral (VUR) de baja presión presente en aproximadamente el 75% de los pacientes. Si bien estas vejigas de gran volumen y complacientes permiten un almacenamiento a baja presión, a menudo presentan vaciamiento incompleto secundario a una contractilidad del detrusor reducida. Por lo tanto, la intervención urológica inicial se orienta al drenaje vesical para evitar la infección del tracto urinario (UTI) y así preservar la función renal. En consecuencia, la circuncisión, los antibióticos profilácticos y la micción programada/doble micción suelen implementarse para disminuir la incidencia de UTI y proteger las vías urinarias superiores de mayor daño.54,55,56,57 Cuando dicho manejo conservador no logra prevenir la infección y conseguir un vaciamiento adecuado, estos niños pueden requerir cistoplastia de reducción y /o apendicovesicostomía para facilitar el cateterismo intermitente limpio y/o cirugía antirreflujo para el manejo del VUR asociado.58,59,60
Pared abdominal
La reconstrucción de la pared abdominal, en la que la porción periférica más normal de la pared abdominal se avanza para sostener esta porción central anómala y deficiente, es una consideración importante en el manejo quirúrgico de los pacientes con PBS. Además del resultado estético, se ha demostrado que la abdominoplastia mejora la dinámica vesical funcional independientemente de la reconstrucción genitourinaria. Las pruebas urodinámicas suelen realizarse en el preoperatorio para determinar la necesidad de procedimientos vesicales concomitantes, como la apendicovesicostomía en casos de vaciamiento incompleto y/o la reimplantación ureteral cuando está presente el VUR. Tradicionalmente, se han descrito en la literatura tres procedimientos quirúrgicos con sus modificaciones para la corrección de los defectos de la pared abdominal asociados con PBS.61,62,63 Si bien el procedimiento de Randolph implica la escisión de una porción de la pared abdominal inferior para corregir la redundancia fascial vertical, los procedimientos de Monfort (Figura 4), (Figura 5), (Figura 6), (Figura 7), (Figura 8) y de Ehrlich emplean la superposición vertical de la fascia para corregir la redundancia lateral junto con el fortalecimiento de la pared abdominal. También se ha descrito un abordaje laparoscópico de la abdominoplastia, que ha demostrado ser beneficioso tanto para asistir en la reconstrucción en sí como para delimitar los distintos grados de deficiencia muscular.64 Asimismo, se ha observado que los pacientes con PBS pueden presentar características únicas con respecto a la variación en la gravedad y el patrón de la deficiencia de la musculatura abdominal, como un área particular con una ausencia completa de músculo. Smith y sus colegas informaron recientemente sus adaptaciones del procedimiento de Monfort, que permiten una corrección más individualizada de la laxitud de la pared abdominal y posicionar el ombligo en una ubicación más anatómicamente correcta.
Figura 4 Una incisión en la línea media desde la apófisis xifoides hasta el pubis circunscribiendo el ombligo.
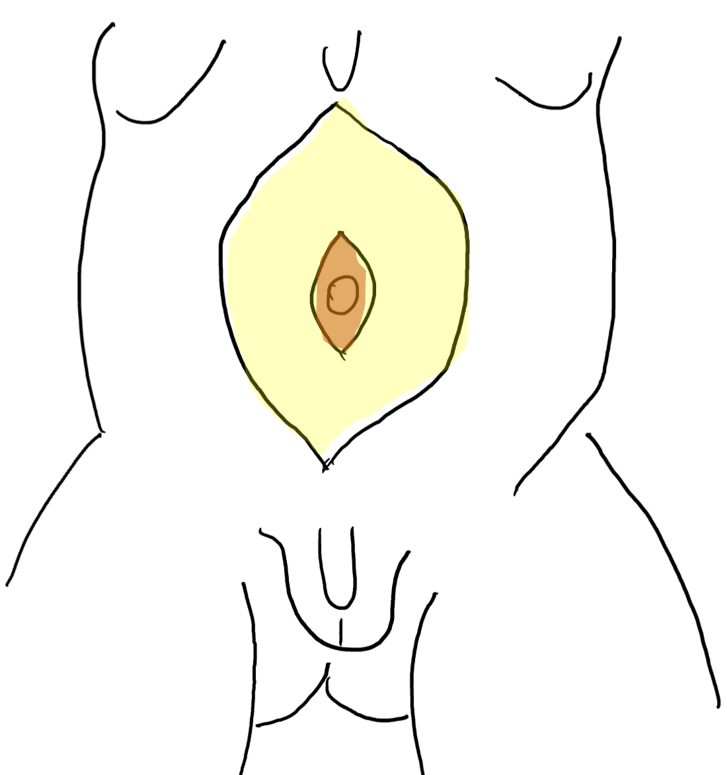
Figura 5 Elevación del colgajo cutáneo entre la grasa subcutánea y el plano fascial. El ombligo está sostenido por la banda fascial central.
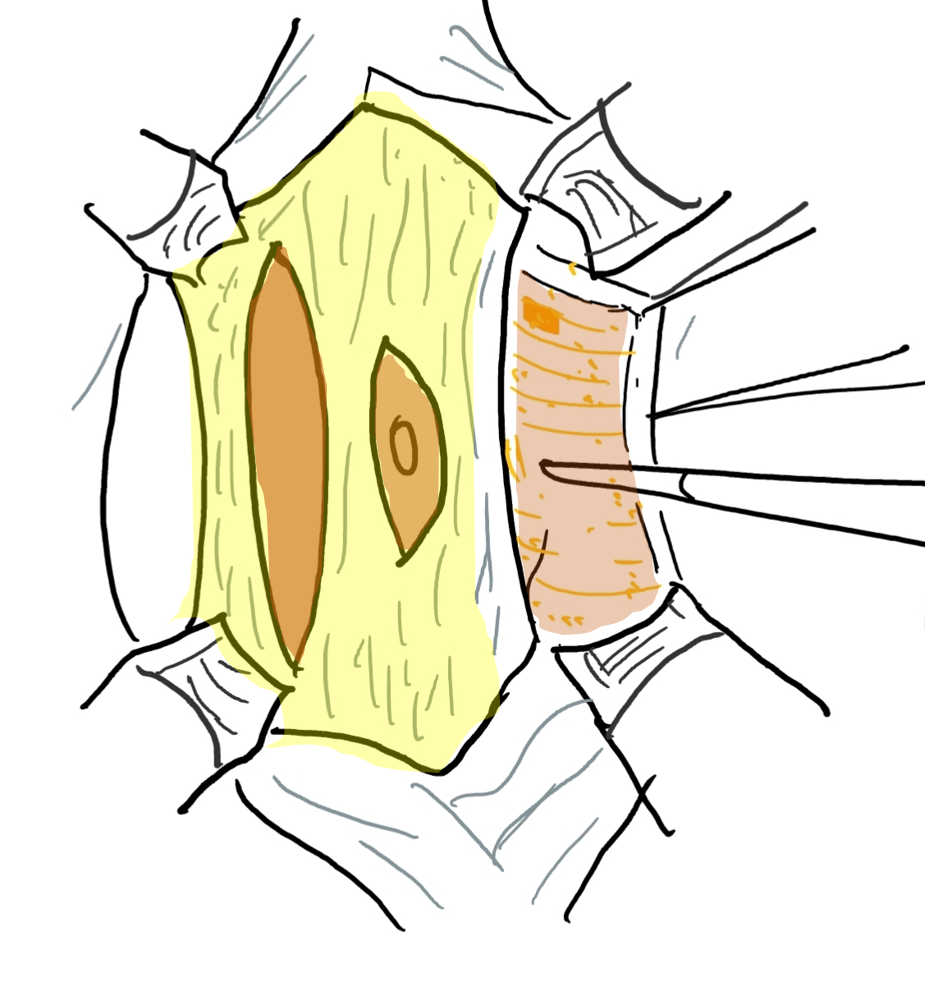
Figura 6 La capa fascial se abre mediante una incisión lateral y paralela al trayecto de las arterias epigástricas superior e inferior. La extensión lateral es la línea axilar anterior.
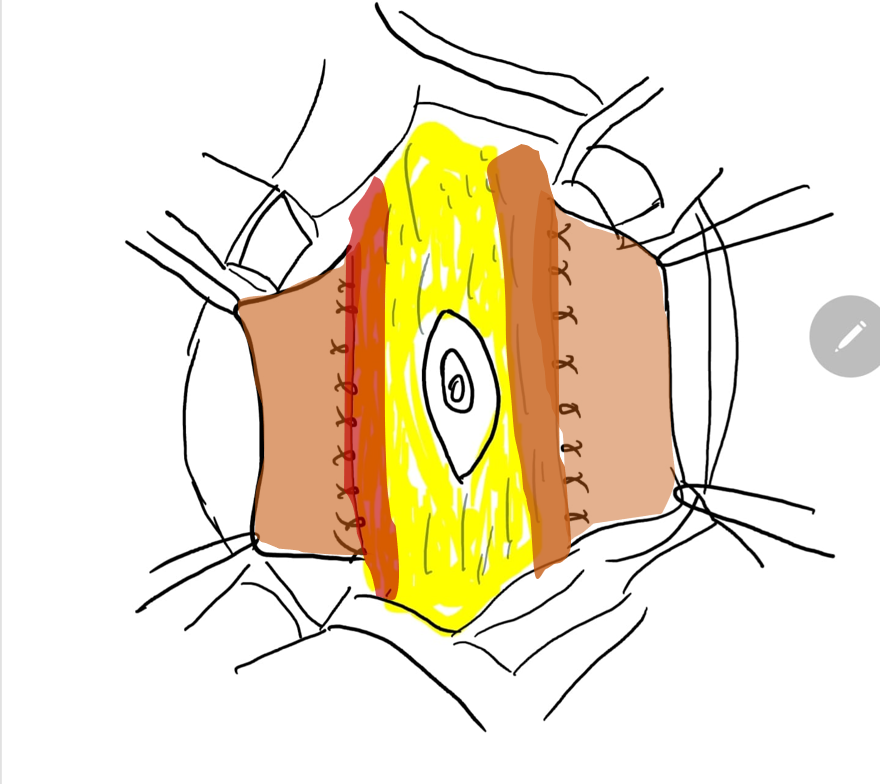
Figura 7 El borde lateral de la tira fascial central se sutura por debajo del músculo.
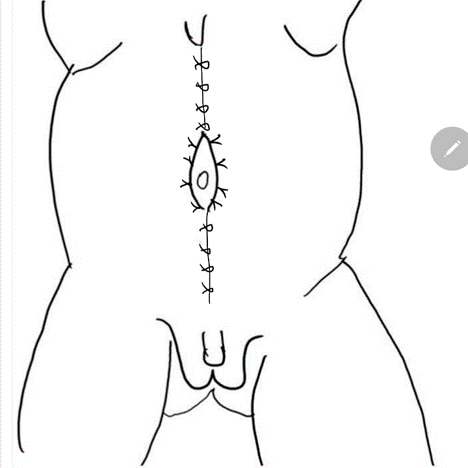
Figura 8 La fascia lateral se fija luego en la línea media por encima y por debajo del ombligo. El cierre pantalón-sobre-chaleco proporciona soporte adicional y los colgajos cutáneos de ambos lados se cierran se cierran posteriormente.
Criptorquidia
Los testículos intraabdominales bilaterales son otra característica distintiva del PBS. La orquidopexia tradicional en un solo tiempo y la orquidopexia de Fowler- Stephens en un solo tiempo versus en etapas son todas opciones potenciales para el manejo de los testículos, según la longitud de los vasos gonadales, y pueden realizarse mediante un abordaje abierto o laparoscópico según la preferencia del cirujano.65,66,67 La pared abdominal laxa puede generar posibles problemas técnicos al crear y mantener el neumoperitoneo, los cuales pueden minimizarse mediante el uso de trócares de expansión radial y altas tasas de flujo de gas durante la orquidopexia laparoscópica, como se describe por Philip et al.67,68 Debe enfatizarse, sin embargo, que, en ausencia de cualquier problema cardiopulmonar predominante, la orquidopexia debe realizarse lo antes posible, tal como recomienda la actual [guía de la AUA](https://www.auanet.org/guidelines/guidelines/cryptorchidism-guideline) y no retrasarse hasta edades mayores de la infancia, a fin de aumentar las probabilidades de fertilidad y mejorar las posibilidades de un procedimiento en un solo tiempo, ya que la división de los vasos gonadales es menos probable cuando la orquidopexia se realiza de forma precoz.
Reconstrucción integral
La reconstrucción integral en el paciente con síndrome de Prune Belly incluye cistoplastia de reducción, resección del uréter distal redundante y reimplante ureteral bilateral con reducción de calibre.69 La reconstrucción puede combinarse con la realización de la abdominoplastia y la orquidopexia bilateral, usualmente antes de los dos años de edad. Este enfoque agresivo se ha utilizado en muchas series y ha mostrado buenos resultados con respecto a una función renal estable o mejorada.
En el abordaje descrito por Woodward, se realiza una amplia incisión abdominal baja que se extiende desde la punta de la 12.ª costilla, a lo largo de la espina ilíaca anterosuperior hasta el pubis y luego se curva hacia arriba hasta la 12.ª costilla contralateral. Esto tiende a preservar la inervación y la vascularización segmentarias de la pared abdominal remanente para la reconstrucción. Además, en el momento de la escisión de la pared abdominal redundante, se preserva la porción con mayor cantidad de fibras musculares. La exposición del tracto urinario que proporciona esta incisión es excepcional. Sin embargo, el uso de esta técnica ha disminuido debido a preocupaciones respecto de la interrupción de los vasos epigástricos y del tejido abdominal lateral redundante. En su lugar, la mayoría de los cirujanos prefieren el abordaje de Monfort (Figura 4), (Figura 5), (Figura 6), (Figura 7), y (Figura 8)
Se incide el peritoneo posterior a lo largo del trayecto de los uréteres. Los vasos espermáticos se movilizan distalmente, teniendo cuidado de no comprometer las inserciones peritoneales del conducto deferente si se requiere una orquidopexia de Fowler-Stephens. La vejiga se abre en la línea media con electrocauterio. Se identifican los orificios ureterales ubicados lateralmente y se moviliza cada uréter. La gran longitud de los uréteres movilizados permite la resección de su tercio distal, que suele ser la porción más anómala. Se realiza una taperización formal o imbricación. Posteriormente, los uréteres se reimplantan, utilizando una técnica transtrigonal o Leadbetter Politano. Se colocan catéteres en los uréteres intervenidos y estos se exteriorizan a través de la vejiga y de incisiones abdominales separadas.
Antes de la cistoplastia de reducción, los testículos se movilizan con un amplio colgajo de peritoneo posterior. En algunos casos, los testículos pueden estar adheridos a la vejiga aumentada de tamaño y deben movilizarse meticulosamente para preservar la vascularización. Si la reconstrucción se realiza antes de los 2 años de edad, puede efectuarse una orquidopexia estándar con vasos espermáticos intactos el 80% de las veces. De lo contrario, será necesaria una orquidopexia de Fowler-Stephens diferida o formal
Se reduce el tamaño de la vejiga mediante la resección del remanente uracal y de la enorme cúpula vesical. Puede resecarse una tira de epitelio vesical de una de las paredes laterales y cerrarse mediante la técnica de chaleco sobre pantalón. La vejiga se drena con un catéter de Malecot. Los testículos previamente movilizados se hacen pasar a través de la pared abdominal distal a nivel del tubérculo púbico. Se crea un bolsillo de dartos en el escroto y cada testículo se fija en el bolsillo con suturas permanentes.
La pared abdominal redundante se mantiene tensa y se marca en la línea media y lateralmente a nivel de los ángulos costovertebrales para delimitar el tejido a resecar. Es fundamental evitar la resección de cantidades excesivas de pared abdominal. La segunda línea de incisión es paralela a la incisión previa. Se reseca una cuña grande de pared abdominal de espesor total. El cierre de la pared abdominal se inicia mediante la colocación de suturas clave de material no absorbible calibre 0 o 1–0 en las espinas ilíacas anteriores y en cada tubérculo púbico. Estas suturas deben abarcar el periostio del hueso y todo el espesor del borde tisular proximal opuesto. La tensión creada ocasionalmente permite la resección de pared abdominal lateral redundante adicional. Cada sección entre las suturas clave se cierra con puntos interrumpidos aproximando todo el espesor del tejido abdominal. El tejido subcutáneo se cierra para evitar espacios muertos y la piel se aproxima con una sutura absorbible subcuticular.
Resultados a largo plazo
Calidad de vida
Aunque el pronóstico global del paciente con PBS en cuanto a supervivencia y calidad de vida ha mejorado considerablemente, la PBS afecta profundamente la calidad de vida relacionada con la salud de los niños afectados, así como la de sus cuidadores.70 Los pacientes presentan un funcionamiento físico, emocional, social y escolar comprometido, y los cuidadores refieren una calidad de vida global inferior debido a las cargas a las que con frecuencia se enfrentan las familias con niños con enfermedades crónicas. Otro estudio demostró que las personas sobrevivientes con PBS tenían una incidencia significativamente alta de afecciones ortopédicas, gastrointestinales y cardiopulmonares que afectaban negativamente su calidad de vida, mientras que el tratamiento de estas afecciones no genitourinarias mejora sus vidas.70,71 Estos estudios subrayan el hecho de que, como responsable principal de la salud global de estos pacientes médicamente complejos, el urólogo pediatra debe estar alerta y preparado no solo para tratar las complicaciones genitourinarias tardías de la PBS, sino también para evaluar y diagnosticar las comorbilidades no genitourinarias que afectan directamente su tratamiento médico y quirúrgico y su calidad de vida, a medida que pasan por la pubertad hacia la edad adulta.
Insuficiencia renal y trasplante
Hasta el 30% de los pacientes, generalmente aquellos con deterioro inicial de la función renal, desarrollan insuficiencia renal crónica durante la infancia o la adolescencia. Se considera que la enfermedad renal terminal (ESRD) de aparición temprana es secundaria a displasia renal, mientras que la insuficiencia renal que aparece más tarde se atribuye con frecuencia al daño parenquimatoso por infecciones repetidas y a la mayor presión transmitida a las vías urinarias superiores generada por el vaciamiento incompleto.72 Yalcinkaya et al informaron que la mediana de edad al inicio de la terapia de reemplazo renal en niños con PBS fue de 7 años, lo que fue significativamente menor que en los pacientes varones con otros tipos de LUTO o displasia renal, con una mediana de edad al primer trasplante de apenas 9,3 años.73
El trasplante renal es necesario en estos pacientes para asegurar un crecimiento y desarrollo normales, y se puede esperar que el éxito del trasplante en pacientes con PBS sea equivalente al de otros grupos de la misma edad. Desde el primer informe en 1976, tanto riñones de donante cadavérico como de donante vivo relacionado se han trasplantado con éxito en pacientes con PBS, con una edad al momento del trasplante que varía de 8 meses a 21 años.73,74 Antes del trasplante, por lo general se realizan nefroureterectomías bilaterales en todos los casos para evitar complicaciones infecciosas. Se recomienda la evaluación radiográfica y urodinámica del tracto urinario inferior para asegurar la ausencia de obstrucción y un vaciamiento vesical equilibrado. El uso de cateterismo intermitente limpio para vaciar la vejiga descompensada no es una contraindicación para el trasplante renal. Todos los pacientes deben mantenerse con profilaxis antibiótica. Una complicación inusual del trasplante en el síndrome de prune belly es la torsión del aloinjerto.75 La torsión, con la consiguiente pérdida del injerto, fue consecuencia de la falta de tono de la pared abdominal. Se ha recomendado la nefropexia para evitar esta complicación desastrosa. En una revisión retrospectiva de ocho pacientes con síndrome de prune belly que se sometieron a trasplante, no hubo diferencias estadísticamente significativas en la mortalidad de los pacientes, la supervivencia del injerto ni la función del injerto en comparación con controles pareados por edad. Un análisis retrospectivo más reciente de una supervivencia del injerto a 10 años del 47% no fue estadísticamente diferente de la de pacientes control pareados por edad,73,74
Función vesical
Como se mencionó antes, los estudios urodinámicos de pacientes con PBS generalmente muestran complacencia normal con sensibilidad y actividad contráctil disminuidas. Como consecuencia, algunos pacientes tienden a retener grandes volúmenes en la vejiga, la cual desarrolla una capacidad progresivamente aumentada asociada a un residuo postmiccional significativo. La falta de soporte abdominal, la eventual asociación de poliuria y la conducta negativa de algunos pacientes respecto a la micción programada suelen aumentar esta tendencia. Por lo tanto, para prevenir mayor dilatación vesical y deterioro del tracto urinario superior, todos los pacientes, incluso aquellos que se sometieron a reconstrucción del tracto urinario inferior y abdominoplastia, deben ser entrenados lo antes posible y reforzados de manera constante para vaciar la vejiga según un horario, eventualmente asociándolo a micción doble o triple y a maniobras de Valsalva o de Credé.76 En caso de micción progresivamente desequilibrada, si tal programa no es factible o resulta ineficaz, la cateterización vesical intermitente es obligatoria, ya sea por vía uretral o a través de un conducto de Mitrofanoff. Con el crecimiento y la mejora de la sensibilidad propioceptiva, así como con el aumento del soporte abdominal, eventualmente se consigue una micción eficaz. En la serie de Lopes et al, el 17.4% de los pacientes sometidos a reconstrucción del tracto urinario requirió cateterización intermitente por vía uretral o a través de un conducto de Mitrofanoff. Raramente, algunos de estos niños recuperan una micción normal con buen vaciamiento vesical.
Crecimiento y desarrollo musculoesquelético
Se puede esperar un crecimiento normal en la mayoría de los pacientes con función renal normal, aunque se observó retraso del crecimiento en ausencia de compromiso renal en un tercio de los pacientes en una serie.34 Las deformidades torácicas, como el pectus excavatum, por lo general no comprometen la función pulmonar en el niño mayor y pueden mostrar cierta mejoría con el crecimiento y el ejercicio. Los pacientes con el síndrome pueden presentar anomalías musculoesqueléticas que afectan la función y la estética, como escoliosis, lordosis y anomalías del tobillo y la cadera que pueden requerir intervención ortopédica temprana. En un estudio reciente, se comunicó a los pacientes mayores y a sus familias que la corrección de los problemas musculoesqueléticos mediante abdominoplastia y/o cirugías ortopédicas era la forma más común en que los proveedores de salud mejoraban la calidad de vida de sus pacientes.
Función sexual y fertilidad
Puede esperarse un patrón normal de desarrollo sexual secundario, ya que la producción hormonal por los testículos se conserva, pero la función sexual puede verse afectada por eyaculación retrógrada. La infertilidad primaria suele ser la norma y, cuando está presente, se cree que se debe a una combinación de anomalías histológicas testiculares con espermatogénesis alterada, defectos estructurales de los conductos y anomalías prostáticas que conducen a eyaculación retrógrada.77 Sin embargo, es posible lograr la paternidad mediante técnicas de reproducción asistida (TRA) especialmente en quienes tuvieron una orquiopexia temprana exitosa, y existen informes de nacidos vivos normales en pacientes adultos con PBS clásica, hecho posible gracias a técnicas de recuperación espermática y a la inyección intracitoplasmática de espermatozoides.78,79,80 También se ha descrito un embarazo normal con parto vaginal asistido en una paciente con el síndrome.81
Conclusiones
Como ocurre con la mayoría de las anomalías congénitas complejas, la clave del manejo del PBS es un enfoque multidisciplinario basado en el trabajo en equipo que proporcione una atención individualizada. Dado el amplio espectro de la enfermedad, el tratamiento del síndrome de prune belly es específico para cada paciente. Cuando la infección y la estasis comprometen el crecimiento o la función renal, se aconseja una intervención quirúrgica agresiva para favorecer el drenaje. Los testículos en estos lactantes se colocan en el escroto de forma temprana para mejorar la fertilidad y esto puede lograrse mediante técnicas laparoscópicas o abiertas. Debido a la condición urodinámica cambiante del tracto urinario inferior y sus efectos sobre la función renal y la infección urinaria, todos los pacientes con síndrome de prune belly requieren una vigilancia urológica cuidadosa de por vida.
Puntos clave
- El síndrome de abdomen en ciruela pasa (PBS) incluye una constelación de anomalías con grados variables de gravedad. Los tres hallazgos principales son deficiencia de la musculatura abdominal, testículos intraabdominales bilaterales y tracto urinario anómalo.
- El tracto urinario se caracteriza por grados variables de hidronefrosis, displasia renal, uréteres dilatados y tortuosos, vejiga aumentada de tamaño y uretra prostática dilatada/ megalouretra.
- Anomalías asociadas adicionales afectan el tracto respiratorio, el tracto gastrointestinal, el sistema cardíaco y el sistema musculoesquelético.
- El determinante más importante de la supervivencia a largo plazo suele ser la gravedad de la anomalía del tracto urinario, en particular, el grado de displasia renal.
- La hidronefrosis no obstructiva es la regla. Es la infección renal, más que la obstrucción, la que representa el mayor riesgo para la función renal.
- La intervención urológica inicial se dirige al drenaje vesical y al vaciamiento adecuado de la vejiga para evitar la infección del tracto urinario (ITU) y así preservar la función renal.
- El manejo quirúrgico consiste en la reconstrucción del tracto urinario, abdominoplastia y orquiopexia bilateral, que pueden realizarse por separado o en una sola etapa en niños más pequeños.
- La atención de los pacientes con PBS requiere un amplio enfoque de equipo multidisciplinario para ayudar a estos niños a prosperar, ganar peso y estar preparados para la cirugía urológica si fuera necesaria.
Referencias
- Woodard S JR, E.A.. Prune belly syndrome.
- Denes FT, Lopes RI. Prune-belly syndrome. In: Partin AW, Dmochowski RR, Kavoussi LR, Peters CA, editors. Campbell wals-wein urology. 12th ed. Philadelphia: Elsevier, Inc; 2021. DOI: 10.25060/residpediatr-2018.v8n1-07.
- Grimsby GM, Harrison SM, Granberg CF, Bernstein IH, Baker LA. Impact and frequency of extra-genitourinary manifestations of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 280 281–286. DOI: 10.1016/j.jpurol.2015.06.005.
- Wong DG, Arevalo MK, Passoni NM, Iqbal NS, Jascur T, Kern AJ. Phenotypic severity scoring system and categorization for prune belly syndrome: application to a pilot cohort of 50 living patients. BJU Int 2019; 123 (1): 130–139. DOI: 10.1111/bju.14524.
- Wheatley JM, Stephens FD, Hutson JM. Prune-belly syndrome: ongoing controversies regarding pathogenesis and management. Semin Pediatr Surg 1996; 5 (2): 95–106.
- Gonzalez R, Reinberg Y, Burke B, Wells T, Vernier RL. Early bladder outlet obstruction in fetal lambs induces rental dysplasia and the prune-belly syndrome. J Pediatr Surg 1990; 25 (3): 342–345. DOI: 10.1016/0022-3468(90)90083-l.
- Ramasamy R, Haviland M, Woodard B JR, J.G.. Patterns of inheritance in familial prune belly syndrome. Urology 2005; 65 (6). DOI: 10.1016/j.urology.2004.12.050.
- Reinberg Y, Shapiro E, Manivel JC, Manley CB, Pettinato G, Gonzalez R. Prune belly syndrome in females: A triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr 1991; 118 (3). DOI: 10.1016/s0022-3476(05)82153-5.
- Balaji KC, Patil A, Townes PL, Primack W, Skare J, Hopkins T. Concordant prune belly syndrome in monozygotic twins. Urology 2000; 55 (6). DOI: 10.1016/s0090-4295(00)00452-0.
- Iqbal NS, Jascur TA, Harrison SM, Edwards AB, Smith LT, Choi ES. Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene. BMC Med Genet 2020; 21 (1). DOI: 10.1186/s12881-020-0973-x.
- Granberg CF, Harrison SM, D D. Genetic basis of prune belly syndrome: Screening for HNF1beta gene. J Urol 2012; 187 (1).
- Stephens FD, Gupta D. Pathogenesis of the prune belly syndrome. J Urol 1994; 152: 2328–2331. DOI: 10.1016/s0022-5347(17)31669-5.
- Palmer JM, Tesluk H. Ureteral pathology in the prune belly syndrome. J Urol 1974; 111 (5). DOI: 10.1016/s0022-5347(17)60050-8.
- Gearhart JP, Lee BR, Partin AW, Epstein JI, Gosling JA, Kogan BA. A quantitative histological evaluation of the dilated ureter of childhood. II: Ectopia, posterior urethral valves and the prune belly syndrome. J Urol 1995; 153 (1). DOI: 10.1097/00005392-199501000-00069.
- Snyder HM, Harrison NW, Whitfield HN, Williams I. Urodynamics in the prune belly syndrome. Br J Urol 1976; 48 (7): 663–670. DOI: 10.1111/j.1464-410x.1976.tb06716.x.
- Kinahan TJ, Churchill BM, McLorie GA, Gilmour RF, Khoury AE. The efficiency of bladder emptying in the prune belly syndrome. J Urol 1992; 148 (2 Pt 2): 600–603. DOI: 10.1016/s0022-5347(17)36665-x.
- Favorito LA, Pires RS, Gallo CM, Sampaio FJB. Study of prostate growth in prune belly syndrome and anencephalic fetuses. J Pediatr Surg 2020; 55 (10): 2221–2225. DOI: 10.1016/j.jpedsurg.2019.10.054.
- Volmar KE, Fritsch MK, Perlman EJ, Hutchins GM. Patterns of congenital lower urinary tract obstructive uropathy: Relation to abnormal prostate and bladder development and the prune belly syndrome. Pediatr Dev Pathol 2001; 4 (5). DOI: 10.1007/s10024001-0042-1.
- Deklerk DP, Scott WW. Prostatic Maldevelopment in the prune belly syndrome: A defect in prostatic stromal-epithelial interaction. J Urol 1978; 120 (3). DOI: 10.1016/s0022-5347(17)57168-2.
- Cabral B, Majidi A, Gonzalez R. Ectopic vasa deferentia in an infant with the prune belly syndrome. J Urol 1988; 94 (4).
- Kroovand RL, Al-Ansari RM, Perlmutter AD. Urethral and genital malformations in prune belly syndrome. J Urol 1982; 127 (1): 94–96. DOI: 10.1016/s0022-3468(82)80571-x.
- Berdon WE, Baker DH, Wigger HJ, Blanc WA. The radiologic and pathologic spectrum of the prune belly syndrome. The importance of urethral obstruction in prognosis. Radiol Clin North Am 1977; 15 (1).
- Hoagland MH, Hutchins GM. Obstructive lesions of the lower urinary tract in the prune belly syndrome. J Urol 1987; 138 (2). DOI: 10.1016/s0022-5347(17)43220-4.
- Volmar KE, Nguyen TC, Holcroft CJ, Blakemore KJ, Hutchins GM. Phimosis as a cause of the prune belly syndrome: Comparison to a more common pattern of proximal penile urethra obstruction. Virchows Arch 2003; 442 (2). DOI: 10.1007/s00428-002-0730-x.
- Beasley S, Bettenay F, Hutson J. The anterior urethra provides clues to the aetiology of prune belly syndrome. Pediatr Surg Int 1988; 3–3(2–3. DOI: 10.1007/bf00182775.
- Sellers BB, McNeal R, Smith RV, Griswold WR, Mendoza SA. Congenital megalourethra associated with prune belly syndrome. J Urol 1976; 116 (6). DOI: 10.1016/s0022-5347(17)59027-8.
- Uehling DT, Zadina SP, Gilbert E. Testicular histology in triad syndrome. Urology 1984; 23 (4). DOI: 10.1016/0090-4295(84)90141-9.
- Massad CA, Cohen MB, Kogan BA, Beckstead JH. Morphology and histochemistry of infant testes in the prune belly syndrome. J Urol 1991; 146 (6): 1598–1600. DOI: 10.1016/s0022-5347(17)38178-8.
- Orvis BR, Bottles K, Kogan BA. Testicular histology in fetuses with the prune belly syndrome and posterior urethral valves. J Urol 1988; 139 (2): 335–337. DOI: 10.1016/s0022-5347(17)42403-7.
- Sayre R, Stephens R, Chonko AM. Prune belly syndrome and retroperitoneal germ cell tumor. Am J Med 1986; 81 (5). DOI: 10.1016/s0022-5347(17)44221-2.
- Woodhouse CR, Ransley PG. Teratoma of the testis in the prune belly syndrome. Br J Urol 1983; 55 (5).
- Mininberg DT, Montoya F, Okada K, Galioto F, Presutti R. Subcellular muscle studies in the prune belly syndrome. J Urol 1973; 109 (3). DOI: 10.1097/00006534-197311000-00052.
- Afifi AK, Rebeiz J, Mire J, Andonian SJ, Kaloustian VM. The myopathology of the prune belly syndrome. J Neurol Sci 1972; 15 (2). DOI: 10.1016/0022-510x(72)90003-2.
- Loder RT, Guiboux JP, Bloom DA, Hensinger RN. Musculoskeletal aspects of prune-belly syndrome. Description and Pathogenesis Am J Dis Child 1992; 146 (10): 1224–1229. DOI: 10.1001/archpedi.1992.02160220110034.
- Geary DF, MacLusky IB, Churchill BM, McLorie G. A broader spectrum of abnormalities in the prune belly syndrome. J Urol 1986; 135 (2). DOI: 10.1016/s0022-5347(17)45627-8.
- Alford BA, Peoples WM, Resnick JS, L’Heureux PR. Pulmonary complications associated with the prune-belly syndrome. Radiology 1978; 129 (2). DOI: 10.1148/129.2.401.
- Wright B JR, RF N, JC P, ET S, ME S, L.E.. Gastrointestinal malformations associated with prune belly syndrome: Three cases and a review of the literature. Pediatr Pathol 1986; 5 (3–4). DOI: 10.3109/15513818609068868.
- Salihu HM, Tchuinguem G, Aliyu MH, Kouam L. Prune belly syndrome and associated malformations. A 13-year experience from a developing country. West Indian Med J 2003; 52 (4).
- Brinker MR, Palutsis RS, Sarwark JF. The orthopaedic manifestations of prune-belly (Eagle-Barrett) syndrome. J Bone Joint Surg 1995; 77 (2). DOI: 10.2106/00004623-199502000-00012.
- Cazorla E, Ruiz F, Abad A, Monleon J. Prune belly syndrome: Early antenatal diagnosis. Eur J Obstet Gynecol Reprod Biol 1997; 72 (1). DOI: 10.1016/s0301-2115(96)02664-4.
- Fitzsimons RB, Keohane C, Galvin J. Prune belly syndrome with ultrasound demonstration of reduction of megacystis in utero. Br J Radiol 1985; 58 (688). DOI: 10.1259/0007-1285-58-688-374.
- Aaronson IA. Posterior urethral valve masquerading as the prune belly syndrome. Br J Urol 1983; 55 (5). DOI: 10.1016/s0022-3468(84)80160-8.
- Tonni G, Ida V, Alessandro V, Bonasoni MP. Prune-belly syndrome: case series and review of the literature regarding early prenatal diagnosis, epidemiology, genetic factors, treatment, and prognosis. Fetal Pediatr Pathol 2013; 31 (1): 13–24. DOI: 10.3109/15513815.2012.659411.
- Yamamoto H, Nishikawa S, Hayashi T, Sagae S, Kudo R. Antenatal diagnosis of prune belly syndrome at 11 weeks of gestation. J Obstet Gynaecol Res 2001; 27 (1). DOI: 10.1111/j.1447-0756.2001.tb01213.x.
- Seidel NE, Arlen AM, Smith EA, Kirsch AJ. Clinical manifestations and management of prune-belly syndrome in a large contemporary pediatric population. Urology 2015; 85 (1). DOI: 10.1016/j.urology.2014.09.029.
- Woods AG, Brandon DH. Prune belly syndrome. A focused physical assessment. Adv Neonatal Care 2007; 7 (3): 144–145.
- Arlen AM, Nawaf C, Kirsch AJ. Prune belly syndrome: current perspectives. Pediatric Health Med Ther 2019; 10: 75–81. DOI: 10.2147/phmt.s188014.
- Rushton HG, Majd M, Chandra R, Yim D. Evaluation of 99M technetium-dimercapto-succinic acid renal scans in experimental acute pyelonephritis in piglets. J Urol 1988; 140 (5). DOI: 10.1016/s0022-5347(17)41992-6.
- Garcia-Roig ML, Grattan-Smith JD, Arlen AM, Smith EA, Kirsch AJ. Detailed evaluation of the upper urinary tract in patients with prune belly syndrome using magnetic resonance urography. J Pediatr Urol 2016; 12 (2). DOI: 10.1016/j.jpurol.2015.11.008.
- Kaplan BS. In utero intervention in prune-belly syndrome. Pediatr Nephrol 1999; 13 (2). DOI: 10.1007/s004670050581.
- Makino Y, Kobayashi H, Kyono K, Oshima K, Kawarabayashi T. Clinical results of fetal obstructive uropathy treated by vesicoamniotic shunting. Urology 2000; 55 (1). DOI: 10.1016/s0090-4295(99)00403-3.
- Burbige KA, Amodio J, Berdon WE, Hensle TW, Blanc W, Lattimer JK. Prune belly syndrome: 35 years of experience. J Urol 1987; 137 (1). DOI: 10.1016/s0022-5347(17)43880-8.
- Fallat ME, Skoog SJ, Belman AB, Eng G, Randolph JG. The prune belly syndrome: A comprehensive approach to management. J Urol 1989; 142 (3). DOI: 10.1016/s0022-5347(17)38895-x.
- Tank ES, McCoy G. Limited surgical intervention in the prune belly syndrome. J Pediatr Surg 1983; 18 (6). DOI: 10.1016/s0022-3468(83)80004-9.
- Woodhouse CRJ, Kellett MJ, Williams DI. Minimal surgical interference in the prune belly syndrome. Br J Urol 1979; 51 (6). DOI: 10.1111/j.1464-410x.1979.tb03582.x.
- Woodard P JR, T.S.. Reconstruction of the urinary tract in prune belly uropathy. J Urol 1978; 119 (6). DOI: 10.1016/s0022-5347(17)57644-2.
- Bukowski TP, Perlmutter AD. Reduction cystoplasty in the prune belly syndrome: a long-term followup. J Urol 1994; 152 (6 Pt 1): 2113–2116. DOI: 10.1016/s0022-5347(17)32333-9.
- Wille MA, Jayram G, Gundeti MS. Feasibility and early outcomes of robotic-assisted laparoscopic Mitrofanoff appendicovesicostomy in patients with prune belly syndrome. BJU Int 2011; 109 (1). DOI: 10.1111/j.1464-410x.2011.10317.x.
- Ehrlich RM, Lesavoy MA, Fine RN. Total abdominal wall reconstruction in the prune belly syndrome. J Urol 1986; 136: 282–285. DOI: 10.1016/s0022-5347(17)44842-7.
- Randolph J, Cavett C, Eng G. Abdominal wall reconstruction in the prune belly syndrome. J Pediatr Surg 1981; 16 (6): 960–964. DOI: 10.1016/s0022-5347(17)52848-7.
- Monfort G, Guys JM, Bocciardi A, Coquet M, Chevallier D. A novel technique for reconstruction of the abdominal wall in the prune belly syndrome. J Urol 1991; 146 (2 Pt 2): 639–640. DOI: 10.1016/s0022-5347(17)37880-1.
- Denes FT, Lopes RI, Oliveira LM, Tavares A, Srougi M. Modified abdominoplasty for patients with the Prune Belly syndrome. Urology 2014; 83 (2): 451–454. DOI: 10.1016/j.urology.2013.09.031.
- Fearon JA, Varkarakis G. Dynamic abdominoplasty for the treatment of prune belly syndrome. Plast Reconstr Surg 2012; 130 (3). DOI: 10.1097/prs.0b013e31825dc170.
- Franco I. Laparoscopic assisted modification of the firlit abdominal wall plication. J Urol 2005; 174 (1). DOI: 10.1097/01.ju.0000161214.65458.c2.
- Patil KK, Duffy PG, Woodhouse CR, Ransley PG. Long-term outcome of Fowler-Stephens orchiopexy in boys with prune belly syndrome. J Urol 2004; 171 (4): 1666–1669. DOI: 10.1097/01.ju.0000118139.28229.f5.
- Yu T-J, Lai M-K, Chen W-F, Wan Y-L. Two-stage orchiopexy with laparoscopic clip ligation of the spermatic vessels in prune-belly syndrome. J Pediatr Surg 1995; 30 (6). DOI: 10.1016/0022-3468(95)90768-8.
- Philip J, Mullassery D, Craigie RJ, Manikandan R, Kenny SE. Laparoscopic orchidopexy in boys with prune belly syndrome—Outcome and technical considerations. J Endourol 2011; 25 (7). DOI: 10.1089/end.2010.0257.
- Saxena AK, Brinkmann OA. Unique features of prune belly syndrome in laparoscopic surgery. J Am Coll Surg 2007; 205 (2). DOI: 10.1016/j.jamcollsurg.2007.03.008.
- Lopes RI, Tavares A, Srougi M, Dénes FT. 27 years of experience with the comprehensive surgical treatment of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 276 271–277. DOI: 10.1016/j.jpurol.2015.05.018.
- Arlen AM, Kirsch SS, Seidel NE, Garcia-Roig M, Smith EA, Kirsch AJ. Health-related quality of life in children with Prune belly Syndrome and their caregivers. Urology 2016; 87 (224). DOI: 10.1016/j.urology.2015.09.028.
- Lesavoy MA, Chang EI, Suliman A, Taylor J, Kim SE, Ehrlich RM. Long-term follow-up of total abdominal wall reconstruction for prune belly syndrome. Plast Reconstr Surg 2012; 129 (1). DOI: 10.1097/prs.0b013e3182362091.
- Reinberg Y, Manivel JC, Pettinato G, Gonzalez R. Development of renal failure in children with the prune belly syndrome. J Urol 1991; 145 (5): 1017–1019. DOI: 10.1016/0022-3468(92)90159-5.
- Alcinkaya F, Bonthuis M, Erdogan BD, Stralen KJ, Baiko S, Chehade H, et al.. Outcomes of renal replacement therapy in boys with prune belly syndrome: findings from the ESPN/ERA-EDTA Registry. Pediatr Nephrol 2018; 33 (1): 117–124. DOI: 10.1007/s00467-017-3770-9.
- Fontaine E, Salomon L, Gagnadoux MF, Niaudet P, Broyer M, Beurton D. Long-term results of renal transplantation in children with the prune-belly syndrome. J 1997; Urol.158(3 Pt 1):892-4. DOI: 10.1097/00005392-199709000-00067.
- Marvin RG, Halff GA, Elshihabi I. Renal allograft torsion associated with prune-belly syndrome. Pediatr Nephrol 1995; 9 (1). DOI: 10.1007/bf00858981.
- Smith CA, Smith EA, Parrott TS, Broecker BH, Woodard JR. Voiding function in patients with the prune-belly syndrome after Monfort abdominoplasty. J Urol 1998; 159 (5). DOI: 10.1016/s0022-3468(99)90298-1.
- Woodhouse CR, Snyder HM. Testicular and sexual function in adults with prune belly syndrome. J Urol 1985; 133 (4): 607 9. DOI: 10.1016/s0022-5347(17)49108-7.
- Halpern JA, Das A, Brannigan RE. Successful sperm retrieval in prune belly syndrome. Asian J Urol 2020; 7 (4): 376–378. DOI: 10.1016/j.ajur.2019.07.004.
- Kolettis PN, Ross JH, Kay R, Thomas AJ. Sperm retrieval and intracytoplasmic sperm injection in patients with prune-belly syndrome. Fertil Steril 1999; 72 (5). DOI: 10.1016/s0015-0282(99)00388-x.
- Fleming SD, Varughese E, Hua VK, Robertson A, Dalzell F, Boothroyd CV. Normal live births after intracytoplasmic sperm injection in a man with the rare condition of Eagle-Barrett syndrome (prune-belly syndrome. Fertil Steril 2013; 100 (6). DOI: 10.1016/j.fertnstert.2013.07.1994.
- Hillman RT, Garabedian MJ, Wallerstein RJ. Pregnancy outcome in a woman with prune belly syndrome. BMJ Case Reports 2012. DOI: 10.1136/bcr-2012-006490.
Última actualización: 2025-09-21 13:35