
23: Atualizações no Manejo da Extrofia
Este capítulo levará aproximadamente 19 minutos para ler.
Introdução
O complexo extrofia-epispádia (EEC) compreende um amplo espectro de anomalias congênitas, todas decorrentes do mesmo defeito embriológico, variando desde epispádia glandular simples até extrofia cloacal.1
Epidemiologia
O complexo extrofia vesical-epispádia-extrofia cloacal (BEEC) é mais frequente em caucasianos. A extrofia vesical clássica (BEX) é o subtipo predominante (50%), ocorrendo em 2.15 - 3.3/100,000 nascidos vivos, e a extrofia cloacal (CEX) é observada em 1/200,000. Nas últimas décadas, houve um aumento na suspeita diagnóstica com os avanços na medicina fetal.2,3
Há predominância do sexo masculino e a razão homem:mulher foi relatada entre 2,3-6:1.4 Entretanto, a extrofia cloacal é a mesma em ambos os sexos (1:1).
A mortalidade em pacientes com extrofia vesical é baixa (4%). Graças aos avanços nas técnicas cirúrgicas e ao manejo atual, a sobrevida a longo prazo desses pacientes é excelente, com relatos de melhorias na fertilidade e na função sexual tanto em mulheres quanto em homens com BEX. Além disso, a sobrevida de crianças com CEX melhorou de 50% em 1960 para mais de 80% atualmente.
Etiopatogênese
Embriologia
A separação da cloaca primitiva em seio urogenital e intestino posterior ocorre durante o primeiro trimestre de gestação, aproximadamente na mesma época em que a parede abdominal anterior é formada. A membrana cloacal é uma camada bilaminar localizada na extremidade caudal do disco germinativo que ocupa a parede abdominal infraumbilical. A interposição do mesoderma entre as camadas ectodérmica e endodérmica da membrana cloacal bilaminar resulta na formação da musculatura abdominal inferior e dos ossos pélvicos. Após ocorrer a interposição mesenquimal, o septo uroretal avança caudalmente e divide a cloaca em bexiga anteriormente e o reto posteriormente. Distalmente, o septo encontra o remanescente posterior da membrana bilaminar, que eventualmente se perfura e forma as aberturas urogenital e anal. Os tubérculos genitais pares migram medialmente e fundem-se na linha média, cefálicos à membrana dorsal, antes da perfuração. Uma falha na migração de células mesenquimais entre as camadas ectodérmica e endodérmica da parede abdominal inferior causa instabilidade da membrana cloacal.5 A ruptura prematura dessa membrana, antes de sua migração caudal, leva ao desenvolvimento desse grupo de anomalias infraumbilicais. A extensão do defeito infraumbilical e o estágio de desenvolvimento no momento em que ocorre a ruptura determinam se resultará em extrofia vesical, extrofia cloacal ou epispádia. Se a ruptura ocorrer após a separação completa dos tratos geniturinário e gastrointestinal, ocorre BEX clássica. No entanto, se isso ocorrer antes da descida do septo uroretal, há externalização do trato urinário inferior e da porção distal do trato gastrointestinal, dando origem à extrofia cloacal (Figura 1).
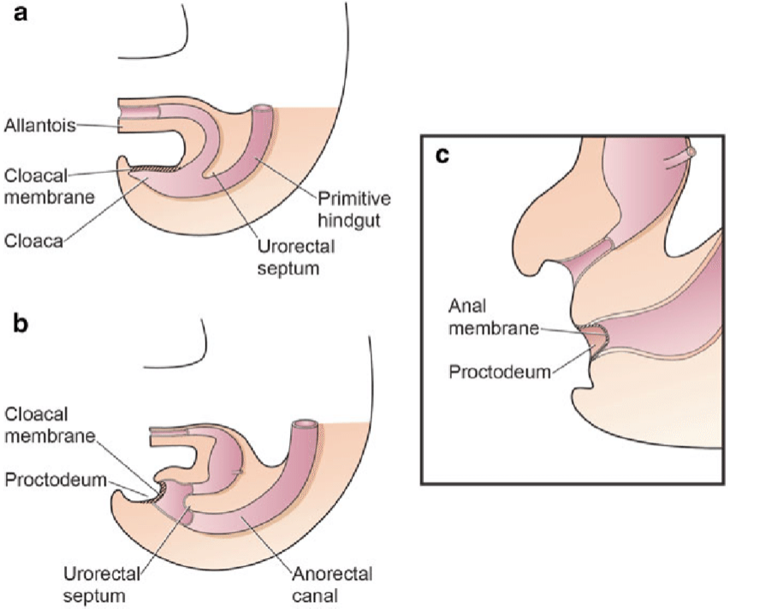
Figura 1 Divisão da cloaca em um seio urogenital primitivo anterior e um reto posterior entre a 4ª e a 6ª semanas de gestação
A teoria mais correlacionada do desenvolvimento embrionário na extrofia, defendida por Marshall e Muecke,6 descreve o defeito básico como um hiperdesenvolvimento inferior anormal da membrana cloacal, que impede a migração medial do tecido mesenquimal. Consequentemente, não ocorre o desenvolvimento adequado da parede abdominal. O momento da ruptura desse defeito cloacal determina a gravidade do distúrbio. Perfurações centrais que resultam em extrofia clássica têm a maior incidência (60%), ao passo que as variantes de extrofia correspondem a 30% e a extrofia cloacal a 10%.
Outras teorias são oferecidas sobre a causa do complexo extrofia-epispádias.7,8 Ambrose e O’Brian9 postularam que um desenvolvimento anormal dos tubérculos genitais, com fusão na linha média abaixo, em vez de acima, da membrana cloacal, resulta no defeito de extrofia. Outra hipótese descreve uma inserção caudal anômala do pedículo corporal, com falha da interposição do tecido mesenquimal na linha média.10 Devido a essa falha, não ocorre a translocação da cloaca para as profundezas da cavidade abdominal. Uma membrana cloacal que permanece em posição infraumbilical superficial representa um estado embrionário instável, com forte tendência à desintegração.11 Nenhuma teoria parece elucidar todos os aspectos do complexo observado clinicamente, e estudos adicionais estão em andamento para descrever completamente o processo de desenvolvimento que, em última instância, forma o complexo extrofia-epispádias.
Etiologia
A etiologia exata desta doença ainda não foi identificada. No entanto, confirmou-se que o evento desencadeador ocorre no início da gestação.8 Vários estudos em andamento sugerem uma possível base genética para o desenvolvimento dessas condições.11 Há uma maior incidência em crianças de mães que receberam grandes doses de progesterona nas fases iniciais da gestação, como em casos de terapias de reprodução assistida, e estima-se que a incidência seja até 7,5 vezes maior nos casos em que foi utilizada a fertilização in vitro.11
Risco de Recorrência
O possível papel dos fatores genéticos implicados na expressão da EEC baseia-se no aumento do risco de recorrência para os descendentes de indivíduos afetados.8 Entre irmãos, Ives et al estimaram o risco de recorrência em aproximadamente 1% em pais de casos de BEX não consanguíneos e não afetados.12 Shapiro et al.13 estimaram o risco de recorrência para irmãos de ascendência europeia para BEX isolada como 1 em 275, tendo pesquisado 2,500 famílias CEB. Também descreveram um risco 400 vezes maior de BEX clássica em descendentes de indivíduos afetados em comparação com a população geral. Com base numa prevalência de 3.3:100,000 (~1:30.000) para BEX isolada entre populações de ascendência europeia, a razão de risco de recorrência entre irmãos foi calculada como 108 (1:275/1:30.000 ~108).14 Embora as pacientes com EEC do sexo feminino representem a minoria dos pacientes, curiosamente, apenas as mulheres afetadas produziram descendentes afetados.13 Entre outras explicações, esse risco de recorrência aparentemente mais elevado para mulheres afetadas pode dever-se a uma maior predisposição genética para EEC (o chamado efeito de Carter).15
Recorrências Familiares
Embora a ocorrência familiar seja rara, 30 famílias multiplex relatadas sustentam a ideia de suscetibilidade genética subjacente à EEC.16 Na maioria dessas famílias, dois membros são afetados. No entanto, em casos raros, a herança da EEC pode ser compatível com herança autossômica dominante com penetrância reduzida ou com um traço autossômico recessivo ou transmissão ligada ao X.17 Essas observações indicam que existem um ou mais genes com efeito maior sobre o fenótipo, embora, na maioria dos casos, fatores causais adicionais sejam necessários para que o fenótipo ocorra. Um pequeno subgrupo de casos pode seguir herança mendeliana, ao passo que, na maioria, a EEC é herdada como um traço complexo, com múltiplos fatores genéticos (herdáveis ou *de novo *mutações somáticas ou de linhagem germinativa), e interações complexas gene-gene, ou gene-ambiente, contribuindo para sua formação.8
Genética Molecular do EEC
As análises citogenéticas e moleculares revelaram anomalias cromossômicas em 20 pacientes com EEC até o momento, embora nenhuma delas pareça ser causal.8 Aberrações cromossômicas numéricas foram observadas em seis pacientes. Em mais quatro pacientes do sexo masculino com BEX, uma paciente do sexo feminino com BEX e uma menina com CEX, foi encontrada uma associação com síndrome de Down.16 A aneuploidia dos cromossomos sexuais em cinco desses casos pode apontar para um lócus gonossômico envolvido na formação da EEC. Aberrações estruturais foram identificadas em seis casos de EEC e em um paciente apresentando simultaneamente CEX e hipomelanose de Ito. Embora os pontos de quebra exatos não tenham sido determinados em nenhum desses casos, foram detectadas várias translocações envolvendo a região q32-ter no cromossomo 9.
Agentes Teratogênicos e EEC
Estudos com gêmeos e dados epidemiológicos sugerem que fatores ambientais desempenham um papel na etiologia da EEC. No entanto, os estudos epidemiológicos existentes não identificaram fatores teratogênicos importantes.4,12,18 Vários estudos confirmaram que o sexo masculino, a raça, a idade parental avançada4 e maior paridade, mesmo após ajuste por idade,19 são fatores de risco predisponentes. Gambhir et al.18 descreveram a exposição materna periconcepcional ao tabagismo como sendo significativamente mais comum em pacientes com CEX do que em um grupo combinado de pacientes com epispádias e BEX clássica. Vários relatos descreveram a ocorrência de EEC em lactentes resultantes de *in vitro *fertilização, mas ainda é objeto de debate se a incidência de crianças com EEC concebidas por *in vitro *fertilização parece ser maior do que o esperado.8,20
Apresentação clínica
Período pré-natal
Apesar da magnitude do defeito na parede abdominal inferior e no desenvolvimento dos órgãos pélvicos, a extrofia vesical ainda é difícil de diagnosticar de forma confiável pela ultrassonografia pré-natal.21 Isso provavelmente se deve à sua baixa incidência e ao fato de ser frequentemente confundida com diagnósticos mais comuns de onfalocele ou gastrosquise.
É possível suspeitar dessas condições durante a gestação quando:
- A bexiga não é identificada em ultrassonografias sucessivas (ausência de enchimento vesical).
- Há diminuição da espessura da parede abdominal.
- Há uma massa abdominal inferior que se torna mais proeminente à medida que a gestação avança.
- Há um cordão umbilical de inserção baixa.
- A genitália apresenta posição anormal (anterior ou posterior) e há dificuldades em determinar o sexo do bebê
- O falo é curto.
- Há aumento do diâmetro pélvico, com separação dos ramos púbicos.
- Há onfalocele.
- Há evidência de anomalias dos membros inferiores e/ou mielomeningocele (sugestiva de extrofia cloacal).
Ultrassonografia tridimensional e o uso crescente de ressonância magnética fetal melhorarão a capacidade de diagnosticar extrofia vesical e cloacal.21 O diagnóstico antenatal permite o aconselhamento pré-natal e a organização do parto em um centro especializado em extrofia. Isso possibilita uma abordagem multidisciplinar por equipes com experiência no manejo da natureza singular do complexo extrofia-epispádias.
Período Neonatal
A maioria das variantes é facilmente identificável ao nascimento.
- A extrofia vesical é mais frequentemente observada em recém-nascidos a termo com bom peso ao nascer.
- Recém-nascidos com extrofia cloacal são geralmente prematuros e pequenos para a idade gestacional.
Infância
- Variantes infrequentes podem passar despercebidas.
- Geralmente são identificadas mais tarde na vida apenas por incontinência urinária persistente ou distúrbios da marcha.
Exame físico
No BEX clássico, a maioria das anomalias consiste em defeitos da parede abdominal, bexiga, genitália, ossos pélvicos e ânus, envolvendo, portanto, o trato urinário inferior, a genitália e o sistema musculoesquelético (membros), enquanto na extrofia cloacal há maior acometimento do trato gastrointestinal e do SNC.
Extrofia Vesical Clássica
A parede abdominal está alongada, e o umbigo é de implantação baixa, localizado na borda superior da placa vesical; pode estar associado a um defeito herniário ou a uma pequena onfalocele. A bexiga está aberta anteriormente, com sua mucosa totalmente exposta; pólipos podem ser vistos na superfície. A urina drena pelos óstios ureterais na superfície da bexiga. O fechamento tardio pode levar a alterações inflamatórias ou mecânicas adicionais, com sinais de inflamação da mucosa, como revestimento esbranquiçado, ulcerações e formações hiperplásicas. As faixas cutâneas finas e brilhantes ao redor da extrofia marcam a transição entre a pele normal e a área de metaplasia escamosa. O ânus é mais anterior, mas a função esfincteriana é normal. Os ossos púbicos estão amplamente separados e também podem estar encurtados e rotados externamente (30%). Eles são palpáveis em ambos os lados do molde da bexiga e na extremidade distal das bordas triangulares. Hérnias inguinais bilaterais são palpáveis na maioria dos pacientes de ambos os sexos.
- BEX feminina: há um clitóris bífido, grandes lábios separados e um mons pubis divergente (Figura 2). A vagina é mais curta do que o normal, não mais de 6 cm de profundidade, mas de calibre normal. O óstio vaginal é frequentemente estenótico e situado anteriormente. Como o ânus também está posicionado ventralmente, o períneo é encurtado.
- BEX masculina: O defeito genital é grave e é provavelmente um dos aspectos mais problemáticos da reconstrução cirúrgica. A placa uretral está aberta e se estende ao longo de um falo curto, largo e curvado dorsalmente, desde a bexiga aberta até o sulco glandular. Ambos os corpos cavernosos estão localizados sob a placa uretral. Exame cuidadoso revela o colículo seminal e os ductos ejaculatórios como minúsculas aberturas na área, onde a próstata está presumivelmente localizada dorsalmente. A glande é aberta e plana (Figura 3), e os testículos de tamanho normal geralmente estão localizados no escroto.
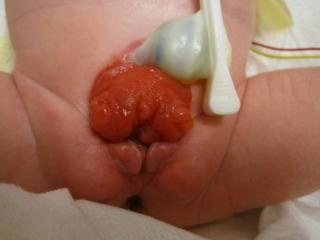
Figura 2 BEX clássico com umbigo de implantação baixa e ânus anteriorizado no períneo. BEX feminino com bexiga aberta e placa uretral, com clitóris bífido.
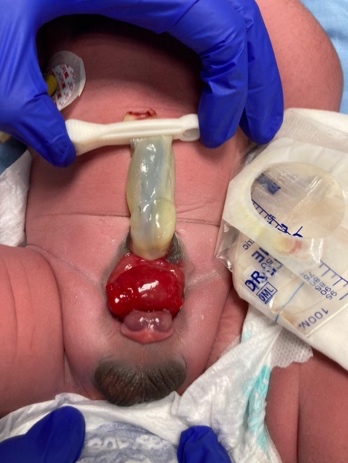
Figura 3 BEX masculino com falo curto, largo e curvado dorsalmente. Distância anormal entre o escroto e o pênis.
Epispádia
O defeito resulta de uma interrupção do desenvolvimento no sentido de não fechamento da placa uretral e, adicionalmente, de uma localização uretral dorsal anormal. Assim, em meninos encontra-se um meato ectópico ou uma faixa mucosa no dorso peniano e, em meninas, detecta-se uma fenda variável da uretra.8 A parede abdominal e os músculos retos, assim como o umbigo, apresentam desenvolvimento completamente normal. A sínfise púbica geralmente está fechada, ou apenas um pequeno hiato está presente, indicando apenas anomalias leves da pelve e do assoalho pélvico. A incontinência urinária parece ser o principal sintoma clínico, dependendo do grau de acometimento do esfíncter urinário. Na maioria dos epispádias distais, não se observa perda involuntária de urina, ao passo que nos casos proximais a urina goteja permanentemente através do meato. Devido às por vezes “menores” alterações clínicas, o epispádias distal pode passar despercebido ao nascimento, especialmente em meninas. Assim, o diagnóstico pode ser feito mais tarde, em idade escolar, devido à incontinência urinária, resistente ao tratamento padrão.
- **Epispádias masculino (Figura 4): De acordo com a localização do meato, o epispádias classifica-se como penopúbico, peniano ou glandular.
- **Epispádias feminino (Figura 5): divide-se em três graus segundo Davis, podendo ser menos grave, com meato patuloso, intermediário ou grave, com fenda envolvendo toda a uretra e o colo vesical, além disso apresentando prolapso da mucosa vesical.
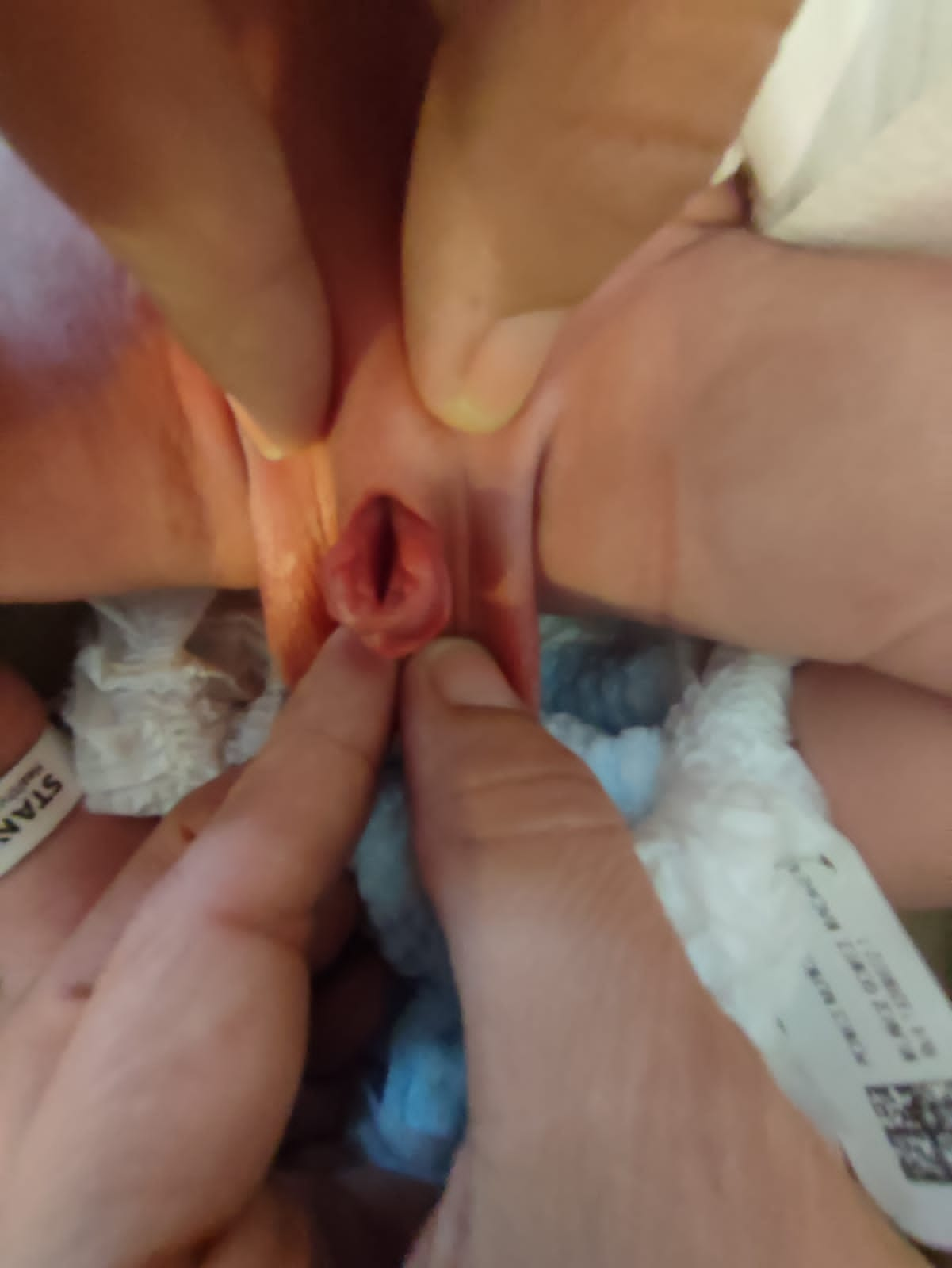
Figura 4 Epispádias masculino
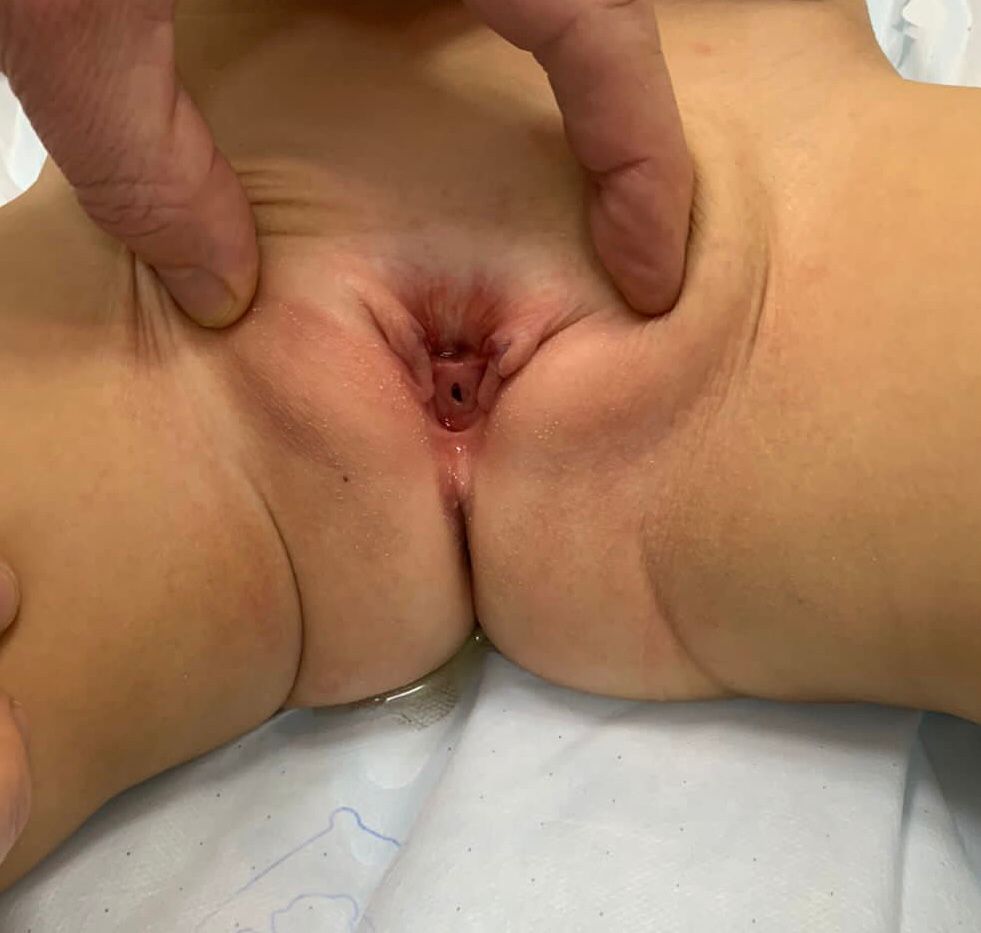
Figura 5 Epispádia feminina
Extrofia cloacal
Os músculos retos abdominais e os ossos púbicos estão separados. A bexiga está aberta na parede abdominal inferior e dividida em 2 metades adjacentes ao segmento exposto do ceco. São evidentes na placa cecal os orifícios que comunicam o íleo terminal, o apêndice (um ou dois) e o intestino distal, e o íleo terminal pode prolapsar como uma ‘tromba’ através dela (aspecto de tromba de elefante). Apresenta-se com ânus imperfurado e pode estar associado a onfalocele. 95% têm mielodisplasia e 65% apresentam uma malformação dos membros inferiores.
-
CEX masculino: o falo é geralmente bífido e pequeno, com cada hemi-glande situada caudal a cada hemi-bexiga, ou pode estar ausente.
-
CEX feminina: o clitóris é bífido e pode haver duas hemivaginas com um útero bicorno (Figura 6).
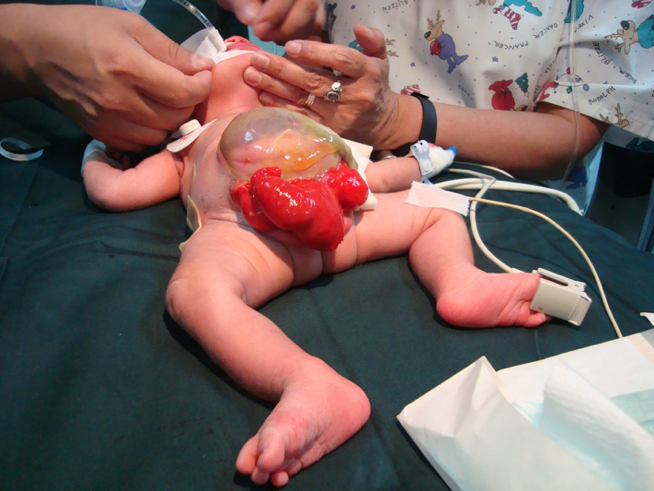
Figura 6 Extrofia cloacal. Onfalocele gigante com 2 hemi-bexigas adjacentes à placa cecal extrofiada. Falo pequeno e bífido, com sua hemi-glande e um hemi-escroto situados distalmente a cada placa vesical.
Variantes da Extrofia
Isto inclui um espectro clinicamente amplo e heterogêneo de anomalias. A extrofia coberta se assemelha à BEX clássica, com parte da mucosa vesical coberta por pele; também pode se apresentar como um epispádias grave com prolapso da bexiga. O umbigo pode estar em posição ortotópica. A pseudoextrofia, no entanto, pode ser muito difícil de identificar após o nascimento e é frequentemente encontrada em idades mais avançadas. A genitália parece normal, e os pacientes podem não apresentar quaisquer sintomas urinários, alguns sendo completamente continentes. No exame físico, pode-se encontrar diástase dos músculos retos abdominais de vários graus. As radiografias pélvicas podem demonstrar uma sínfise púbica aberta, embora não seja incomum que isso seja um achado incidental.
Anomalias Associadas
Anomalias Urológicas
Várias malformações urológicas estão presentes em cerca de um terço de todos os casos de EEC, predominantemente na população com EC (obstrução da junção ureteropélvica, rim pélvico ectópico, rim em ferradura, agenesia renal, megaureter, ectopia ureteral e ureterocele).1,8 Todos os pacientes devem ser submetidos a um procedimento antirrefluxo em cada plastia do colo vesical, pois há prevalência de 100% de refluxo vesicoureteral bilateral devido a uma falha de desenvolvimento da junção ureterovesical em todo o espectro da EEC.
Anomalias Espinhais e Ortopédicas
A incidência de anomalias espinhais varia dentro do espectro EEC. Em crianças nascidas com CEB, as anomalias espinhais ocorrem em cerca de 7% dos casos, ao passo que um grupo heterogêneo de anomalias espinhais congênitas, resultantes do fechamento defeituoso do tubo neural no início da vida fetal e do desenvolvimento anômalo da massa celular caudal, pode ser confirmado por ressonância magnética (RM), sendo identificado em quase 100% dos pacientes com CEX.8 Deve-se ter em mente um componente neurológico nesses pacientes no que diz respeito à função vesical e dos membros inferiores, e à capacidade erétil.22,23 Anomalias esqueléticas e dos membros (deformidades em pé torto congênito, ausência de pés, deformidades tibiais ou fibulares e luxações do quadril) são mais comumente observadas em CEX.1,8 No entanto, não há relatos sobre displasia do quadril no seguimento de longo prazo do EEC.
Anomalias Gastrointestinais
Estas estão predominantemente associadas à CEX. Além de um remanescente comum do intestino posterior de tamanho variável, onfaloceles são encontradas em 88-100% dos casos de CEX. Mal rotação gastrointestinal ou duplicação, assim como a síndrome do intestino curto, podem ser observadas em até 46% dos casos.1,8 Em cerca de 25% dos casos, uma síndrome do intestino curto, seja anatômica ou funcional, causa disfunção absortiva.
Anomalias ginecológicas
Além da genitália externa anormal, o colo do útero apresenta inserção baixa na parede vaginal superior, próxima ao intróito, na maioria dos casos.1,8,24 Apesar disso, a anatomia e a função do útero e dos anexos são normais. O assoalho pélvico e o defeito do levantador do ânus, juntamente com a ausência dos ligamentos cardinais, predispõem as mulheres ao prolapso vaginal ou uterino em cerca de 50% dos casos. As anomalias müllerianas são bastante comuns na CEX, ou seja, duplicação da vagina e do útero, agenesia vaginal.1,8
Estudos de Imagem
Geralmente são realizados para identificar e diagnosticar possíveis anomalias associadas (malformações pélvicas e abdominais, anomalias da coluna e da medula espinhal, etc).
Os principais exames são ultrassonografia renal, ultrassonografia da medula espinhal e radiografias da coluna vertebral e da pelve. Todos os recém-nascidos com CEX devem realizar ultrassonografia da coluna vertebral e radiografias para definir as anomalias espinhais individuais, variando de hemivértebra a mielomeningocele. A ressonância magnética é adicionalmente recomendada no seguimento para identificar anomalias espinhais ocultas que predisponham ao ancoramento sintomático da medula espinhal.
A avaliação ultrassonográfica das articulações do quadril é de importância fundamental para todos os pacientes com EEC. A radiografia pélvica simples pode ser útil para estimar a dimensão da diástase da sínfise púbica e a localização dos quadris.
Diagnóstico
No período antenatal, a EEC pode ser suspeitada a partir da 16ª semana de gestação. Com os avanços na sensibilidade da ultrassonografia antenatal, tem havido um aumento na suspeita pré-natal.
Na maioria dos casos, a malformação é diagnosticada ao nascimento, pois torna-se clinicamente evidente.
Manejo
Se houver diagnóstico ou suspeita pré-natal de EEC, o ideal é aconselhar os pais e o parto deve ser programado em um centro especializado (encaminhar se necessário). Não há experiência que sustente a indicação de cirurgia fetal. O parto vaginal não é recomendado devido ao maior risco de lesão da placa vesical.
Atualmente, isso já não é considerado uma emergência e/ou uma emergência cirúrgica, permitindo a transferência em condições seguras para centros especializados capazes de cuidar dessas crianças com malformações de baixa frequência e altamente complexas.
A abordagem cirúrgica inicialmente concentrou-se na derivação urinária para preservar a função renal. No entanto, desde que o primeiro fechamento vesical bem-sucedido foi relatado por Young em 1942,1,8,11,24,25 avanços significativos foram alcançados nas áreas de reconstrução em estágios, na aparência dos genitais externos e na preservação da continência e da função renal. Com melhorias na técnica cirúrgica, bem como nos cuidados perioperatórios, agora pode-se esperar excelente sobrevida a longo prazo.
Cirurgia
Os objetivos da abordagem cirúrgica atual são;1 reconstrução da parede abdominal,2 realocação e fechamento anatômico da bexiga extrofiada,3 preservando a função renal e alcançando continência urinária, e4 reconstrução da genitália externa.26,27,28,29,30 Historicamente, a reconstrução era realizada em 3 etapas começando no período neonatal. No entanto, atualmente existem centros superespecializados ou equipes multicêntricas que manejam a condição e podem organizar manejo diferido, a fim de oferecer o melhor a cada paciente, acumulando experiência em patologias de baixa frequência e alta complexidade.31,32,33,34,35,36,37,38,39,40
As etapas cirúrgicas são
- Fechamento da bexiga, com aproximação dos ossos púbicos, sem necessidade de osteotomias. Em pacientes com CEX, deve ser realizada derivação intestinal.
- A segunda etapa consiste na reconstrução genital completa. Em alguns centros, também se realiza o Procedimento de Kelly ou uma Reconstrução Radical de Tecidos Moles, para alcançar continência vesical e um maior comprimento peniano nos homens.
- Por fim, a continência urinária deve ser avaliada a médio e longo prazo. Para os casos em que esse objetivo ainda não tenha sido alcançado ou quando houver risco de comprometimento da função renal, há uma terceira etapa na qual podem ser realizados cirurgia do colo vesical e/ou cistoplastia de aumento com uma derivação urinária continente para cateterismo intermitente limpo.
Em diversos centros superespecializados em todo o mundo, optou-se pelo fecho tardio aos dois a quatro meses de idade, realizando uma abordagem em tempo único, juntamente com a cirurgia descrita por Kelly,26,30,39 que inclui:1 dissecção e fecho vesical com ou sem procedimento antirrefluxo (se o tamanho da bexiga e as características da parede o permitirem);2 a mobilização radical dos tecidos moles com desinserção dos ramos do púbis;3 a criação de um colo vesical ou de um mecanismo de continência com o auxílio do estimulador de Peña para identificar o complexo do neoesfíncter muscular e4 reconstrução peniana ou introitoplastia.
Os benefícios dessa abordagem são que se evita a cirurgia prévia no período neonatal, permitindo a preservação dos tecidos nativos e evitando a formação de cicatrizes e fibrose geradas pelo fechamento primário. Isso também favorece o apego parental e o desenvolvimento de um vínculo entre a criança e seus pais, sem a necessidade de uma internação prolongada nos primeiros dias de vida.
Foi demonstrado que o fechamento tardio da bexiga pode ser realizado de forma segura e bem-sucedida e que, em princípio, não afeta o desenvolvimento da bexiga. Adiar a cirurgia permite a maturação do recém-nascido, com a consequente redução dos riscos de uma anestesia prolongada e favorece o crescimento do lactente durante o período de mini-puberdade, além de promover a preparação da equipe de especialistas para cada caso.39
Complicações
Devido à sua complexidade e à frequência muito baixa, essas condições apresentam uma série de complicações principalmente associadas à cirurgia reconstrutiva:34,39
- Precoce: deiscência da ferida cirúrgica, prolapso vesical, fístulas uretrais ou vesicocutâneas (4 – 19%), estenose uretral (8%), isquemia peniana.
- Tardio: RVU, ITUs recorrentes, esvaziamento vesical incompleto, incontinência urinária, litíase vesical, fibrose da parede abdominal, ruptura vesical, prolapso uterino, insuficiência renal, ejaculação retrógrada, oligoespermia e infertilidade, e síndrome do intestino curto e incontinência fecal (extrofia cloacal).
Prognóstico
Se esses pacientes não forem tratados, sofreriam de infecções do trato urinário recorrentes, incontinência urinária permanente e problemas com a atividade sexual, além de apresentarem maior risco de câncer de bexiga, o que, em conjunto, significa uma pior qualidade de vida.26,27,28,29,30,31,32,33,34,35
No entanto, se o manejo adequado da condição for realizado conforme sugerido, esses pacientes têm uma expectativa de qualidade de vida que é provavelmente semelhante à da população geral. Estima-se também que eles potencialmente serão capazes de levar uma vida absolutamente normal, considerando que em mais de 90% dos casos não há associação com outras malformações congênitas. Além disso, esses pacientes poderiam desenvolver continência urinária sem necessitar de aumento vesical ou cateterismo intermitente para esvaziamento, mesmo que a taxa de sucesso seja reservada (apenas 23% de micção voluntária pela uretra com a técnica MSRE,41 39% com a técnica CPRE,28 e 53% com a técnica de Kelly26).
Tabela 1 Ampla variação da taxa de continência das diferentes abordagens, dependendo da definição de continência e do período de observação.42
| Abordagem | Taxa de continência (%) | Literatura |
|---|---|---|
| MSRE | 74 | Gearhart et al |
| 62 | Gupta et al | |
| 22 | Dickson et al | |
| CPRE | 80 | Grady et al |
| 74 | Hammouda et al | |
| 23 | Arab et al | |
| RSTM | 73 | Kelly et al |
| 70 | Jarzebowski et al | |
| 33–67 (feminino) | Cuckow et al | |
| 44–81 (masculino) |
Atualmente, esses pacientes apresentam excelente sobrevida a longo prazo, alcançando continência urinária em até 75-80% dos pacientes com extrofia vesical e 65-70% nos casos de extrofia cloacal;39,43,41 no entanto, muitos deles necessitam de derivações urinárias e intestinais permanentes. Em geral, a função sexual é preservada, e a maioria dos pacientes é fértil. Recomenda-se parto por cesariana em pacientes do sexo feminino que foram submetidas à cirurgia para evitar danos aos mecanismos de continência.
Referências
- Gearhart JP, Gearhart JP, Rink RC. The Bladder Exstrophy–epispadias–cloacal Exstrophy Complex. J Pediatr Urol 2001: 386–415. DOI: 10.1016/b978-1-4160-3204-5.00030-x.
- Siffel C, Correa A, Amar E, Bakker MK, Bermejo-Sánchez E, Bianca S, et al.. Bladder exstrophy: An epidemiologic study from the International Clearinghouse for Birth Defects Surveillance and Research, and an overview of the literature. Am J Med Genet C Semin Med Genet 2011; 157 (4): 321–332. DOI: 10.1002/ajmg.c.30316.
- Cervellione RM, Mantovani A, Gearhart J, Bogaert G, Gobet R, Caione P, et al.. Prospective study on the incidence of bladder/cloacal exstrophy and epispadias in Europe. J Pediatr Urol 2015; 11 (6): 337.e1–337.e6. DOI: 10.1016/j.jpurol.2015.03.023.
- Boyadjiev SA, Dodson JL, Radford CL, Ashrafi GH, Beaty TH, Mathews RI, et al.. Clinical and molecular characterization of the bladder exstrophy-epispadias complex: analysis of 232 families. BJU Int. 2004;94:1337-1343. . DOI: 10.1111/j.1464-410X.2004.05170.x..
- Männer J, Kluth D. The morphogenesis of the exstrophy-epispadias complex: a new concept based on observations made in early embryonic cases of cloacal exstrophy. Anat Embryol (Berl) 2005; 210 (1): 51–57. DOI: 10.1007/s00429-005-0008-6.
- Marshall VF, Muecke EC. Variations in exstrophy of the bladder. Plast Reconstr Surg 1962; 31 (4): 396. DOI: 10.1097/00006534-196304000-00030.
- Stephens FD, Hutson JM. Differences in embryogenesis of epispadias, exstrophy–epispadias complex and hypospadias. J Pediatr Urol 2005; 1 (4): 283–288. DOI: 10.1016/j.jpurol.2005.01.008.
- Ebert AK, Reutter H, Ludwig M, Rösch WH. Exstrophy-epispadias complex. Definitions 2009; 4 (23). DOI: 10.32388/7eqi40.
- Ambrose SS, O’Brien DP. Surgical Embryology of the Exstrophy-Epispadias Complex. Surg Clin North Am 1974; 54 (6): 1379–1390. DOI: 10.1016/s0039-6109(16)40493-7.
- Mildenberger H, Kluth D, Dziuba M. Embryology of bladder exstrophy. J Pediatr Surg. 1988;23:166-170. . DOI: 10.1016/s0022-3468(88)80150-7..
- Gearhart JP. Exstrophy-Epispadias Complex. Campbell-Walsh-Wein Urology, vol. 31. 12th ed. 2021. DOI: 10.1016/b978-1-4160-6911-9.00124-9.
- Ives E, Coffey R, Carter CO. A family study of bladder exstrophy. J Med Genet. 1980;17:139-141. . DOI: 10.1136/jmg.17.2.139..
- Shapiro E, Lepor H, Jeffs RD. The inheritance of the exstrophy-epispadias complex. J Urol. 1984;132:308-310. .
- Reutter H, Qi L, Gearhart JP, Boemers T, Ebert AK, Rösch WH, et al.. Concordance analyses of twins with bladder exstrophy-epispadias complex suggest genetic etiology. Am J Med Genet A. 2007;143:2751-2756. . DOI: 10.1002/ajmg.a.31975.
- Carter CO. Genetics of common single malformations. Br Med Bull. 1976;32:21-26. .
- Ludwig M, Ching B, Reutter H, Boyadjiev SA. The bladder exstrophy-epispadias complex. Birth Defects Res Part A Clin Mol Teratol. 2009;85:509-22. . DOI: 10.1002/bdra.20557..
- Reutter H, Shapiro E, Gruen JR. Seven new cases of familial isolated bladder exstrophy and epispadias complex (BEEC) and review of the literature. Am J Med Genet A. 2003;120A:215-221. . DOI: 10.1002/ajmg.a.20057..
- Gambhir L, Höller T, Müller M, Schott G, Vogt H, Detlefsen B, et al.. Epidemiological survey of 214 European families with Bladder Exstrophy-Epispadias Complex (BEEC) J Urol. 2008;179:1539-1543. . DOI: 10.1016/j.juro.2007.11.092..
- Byron-Scott R, Haan E, Chan A, Bower C, Scott H, Clark K. A population-based study of abdominal wall defects in South Australia and Western Australia. Paediatr Perinat Epidemiol. 1998;12:136-151. . DOI: 10.1046/j.1365-3016.1998.00090.x..
- Wood HM, Babineau D, Gearhart JP. In vitro fertilization and the cloacal/bladder exstrophy-epispadias complex: A continuing association. J Pediatr Urol. 2007;3:305-310. . DOI: 10.1016/j.jpurol.2006.10.007..
- Palmer B, Frimberger D, Kropp B. Bladder exstrophy-epispadias complex and cloacal exstrophy. Section 7.D: Developmental anomalies. Paediatric Urology Book. Paediatric Urology Department, University of Oklahoma; 2011. DOI: 10.1007/978-1-84882-132-3\\_45.
- Rösch WH, Hanisch E, Hagemann M, Neuhuber WL. The Characteristic innervation pattern of the urinary bladder in particular forms of exstrophy-epispadias-complex. BJU. 2001;87:30. .
- Schober JM, Carmichael PA, Hines M, Ransley PG. The ultimate challenge of cloacal exstrophy. J Urol. 2002;167:300-304. . DOI: 10.1016/s0022-5347(05)65455-9..
- Woodhouse CRJ, Hinsch R. The anatomy and reconstruction of the adult female genitalia in classical exstrophy. BJU. 1997;79:618-622. . DOI: 10.1046/j.1464-410X.1997.00148.x..
- Woodhouse. C.R.J.: Genitoplasty in exstrophy and epispadias. Cambridge University Press; 2006, DOI: 10.1017/cbo9780511545757.046.
- Kelly JH. Vesical exstrophy: repair using radical mobilisation of soft tissues. Pediatr Surg Int 1995; 10 (5-6): 298–304. DOI: 10.1007/bf00182207.
- Grady RW, Mitchell ME. Complete primary repair of exstrophy. J Urol. 1999; 62 (4): 415–1420. DOI: 10.1097/00005392-199910000-00071.
- Groth. Bladder exstrophy consortium (MIBEC) after 5 years. AUA Chicago; 2019.
- Dickson AP. The management of bladder exstrophy: the Manchester experience. J Pediatr Surg. 2014; 9 (2): 44–250. DOI: 10.1016/j.jpedsurg.2013.11.031.
- Cuckow P, López PJ. Bladder Exstrophy Closure and Epispadias. In: Spitz L, Coran A, editors. En Operative Pediatric Surgery. 7th ed. 2013. DOI: 10.1201/b13237-101.
- Borer JG, Gargollo PC, Hendren WH, Diamond DA, Peters CA, Atala A, et al.. Early Outcome Following Complete Primary Repair Of Bladder Exstrophy In The Newborn. J Urol 2005; 174 (4 Part 2): 1674–1679. DOI: 10.1097/01.ju.0000175942.27201.59.
- Baird AD, Nelson CP, Gearhart JP. Modern staged repair of bladder exstrophy: a contemporary series. J Pediatr Urol 2007. 4: 11–315. DOI: 10.1016/j.jpurol.2006.09.009.
- Borer JG, Vasquez E, Canning DA, Kryger JV, Mitchell ME. An initial report of a novel multi-institutional bladder exstrophy consortium: a collaboration focused on primary surgery and subsequent care. J Urol. 2015. DOI: 10.1016/j.juro.2014.10.114.
- Ellison JS, Shnorhavorian M, Willihnganz-Lawson K, Grady R, Merguerian PA. A critical appraisal of continence in bladder exstrophy: Long-term outcomes of the complete primary repair. J Pediatr Urol 2016. 2 (4): 05 1–205 2057. DOI: 10.1016/j.jpurol.2016.04.005.
- Schaeffer AJ, Stec AA, Purves JT, Cervellione RM, Nelson CP, Gearhart JP. Complete primary repair of bladder exstrophy: a single institution referral experience. J Urol. 2011; 86 (3): 041–1046. DOI: 10.1016/j.juro.2011.04.099.
- Pathak P, Ring JD, Delfino KR, Dynda DI, Mathews RI. Complete primary repair of bladder exstrophy: a systematic review. J Pediatr Urol. 2020; 6 (2): 49–153. DOI: 10.1016/j.jpurol.2020.01.004.
- Ahn JJ, Shnorhavorian M, Katz C, Goldin AB, Merguerian PA. Early versus delayed closure of bladder exstrophy: A National Surgical Quality Improvement Program Pediatric analysis. J Pediatr Urol. 2018; 4 (1): 7 1–27 5. DOI: 10.1016/j.jpurol.2017.11.008.
- Leclair MD, Villemagne T, Faraj S, Suply E. The radical soft-tissue mobilization (Kelly repair) for bladder exstrophy. J Pediatr Urol. 2015; 1 (6): 64–365. DOI: 10.1016/j.jpurol.2015.08.007.
- Leclair MD, Faraj S, Sultan S. One-stage combined delayed bladder closure with Kelly radical soft-tissue mobilization in bladder exstrophy: preliminary results. J Pediatr Urol. 2018; 4 (6): 58–564. DOI: 10.1016/j.jpurol.2018.07.013.
- Baradaran N, Stec AA, Schaeffer AJ, Gearhart JP, Mathews RI. Delayed primary closure of bladder exstrophy: immediate postoperative management leading to successful outcomes. Urology. 2012; 9 (2): 15–419. DOI: 10.1016/j.urology.2011.08.077.
- Jarzebowski AC, McMullin ND, Grover S SR, BR H, J.M.. The Kelly technique of bladder exstrophy repair: continence, cosmesis and pelvic organ prolapse outcomes. J Urol. 2009. DOI: 10.1016/j.juro.2009.02.083.
- Cervellione RM, Husmann DA, Bivalacqua TJ, Sponseller PD, Gearhart JP. Penile ischemic injury in the exstrophy/epispadias spectrum: new insights and possible mechanisms. J Pediatr Urol. 2010; 5: 50–456. DOI: 10.1016/j.jpurol.2010.04.007.
- Purves JT, Gearhart JP. Complications of radical soft-tissue mobilization procedure as a primary closure of exstrophy. J Pediatr Urol 2008. 1: 5–69. DOI: 10.1016/j.jpurol.2007.02.006.
- Maruf M, Manyevitch R, Michaud J. Urinary Continence Outcomes in Classic Bladder Exstrophy: A Long-Term Perspective. J Urol. 2020; 03 (1): 00–205. DOI: 10.1097/ju.0000000000000505.
- Promm M, Roesch WH. Recent Trends in the Management of Bladder Exstrophy: The Gordian Knot Has Not Yet Been Cut. Front Pediatr 2019; 7. DOI: 10.3389/fped.2019.00110.
Ultima atualização: 2025-09-21 13:35