
23: Actualizaciones en el manejo de la extrofia
Este capítulo durará aproximadamente 20 minutos para leer.
Introducción
El complejo extrofia-epispadias (EEC) comprende un amplio espectro de anomalías congénitas, todas originadas a partir del mismo defecto embriológico, que va desde el epispadias del glande simple hasta la extrofia cloacal.1
Epidemiología
El complejo de extrofia vesical-epispadias-extrofia cloacal (BEEC) es más frecuente en caucásicos. La extrofia vesical clásica (BEX) es el subtipo predominante (50%), presentándose en 2.15 - 3.3/100,000 nacidos vivos, y la extrofia cloacal (CEX) se observa en 1/200,000. En las últimas décadas ha habido un aumento de la sospecha diagnóstica con los avances en la medicina fetal.2,3
Existe un predominio masculino y se ha reportado que la relación varón:mujer está entre 2,3 y 6:1.4 Sin embargo, la exstrofia cloacal es igual en ambos sexos (1:1).
La mortalidad en los pacientes con extrofia vesical es baja (4%). Gracias a los avances en las técnicas quirúrgicas y al manejo actual, la supervivencia a largo plazo de estos pacientes es excelente, registrándose mejoras en la fertilidad y la función sexual tanto en mujeres como en hombres con BEX. Además, la supervivencia de los niños con CEX ha mejorado del 50% en 1960 a más del 80% en la actualidad.
Etiopatogenia
Embriología
La separación de la cloaca primitiva en el seno urogenital y el intestino posterior ocurre durante el primer trimestre del embarazo, aproximadamente al mismo tiempo en que se forma la pared abdominal anterior. La membrana cloacal es una capa bilaminar localizada en el extremo caudal del disco germinativo que ocupa la pared abdominal infraumbilical. La interposición mesodérmica entre las capas ectodérmica y endodérmica de la membrana cloacal bilaminar da lugar a la formación de la musculatura abdominal inferior y los huesos pélvicos. Tras producirse la interposición mesenquimatosa, el tabique urorectal avanza caudalmente y divide la cloaca en la vejiga por delante y el recto por detrás. Distalmente, el tabique se encuentra con el remanente posterior de la membrana bilaminar, que finalmente se perfora y forma los orificios urogenital y anal. Los tubérculos genitales pares migran medialmente y se fusionan en la línea media, cefálicos a la membrana dorsal antes de la perforación. Una falla en la migración de células mesenquimatosas entre las capas ectodérmica y endodérmica de la pared abdominal inferior causa inestabilidad de la membrana cloacal.5 La ruptura prematura de esta membrana, antes de su migración caudal, conduce al desarrollo de este grupo de anomalías infraumbilicales. La extensión del defecto infraumbilical y el estadio de desarrollo en el que ocurre la ruptura determinan si resulta una extrofia vesical, una extrofia cloacal o un epispadias. Si la ruptura ocurre después de la separación completa de los tractos genitourinario y gastrointestinal, ocurre la BEX clásica. Sin embargo, si esto ocurre antes del descenso del tabique urorectal, hay externalización del tracto urinario inferior y de la porción distal del tracto gastrointestinal, dando lugar a extrofia cloacal (Figura 1).
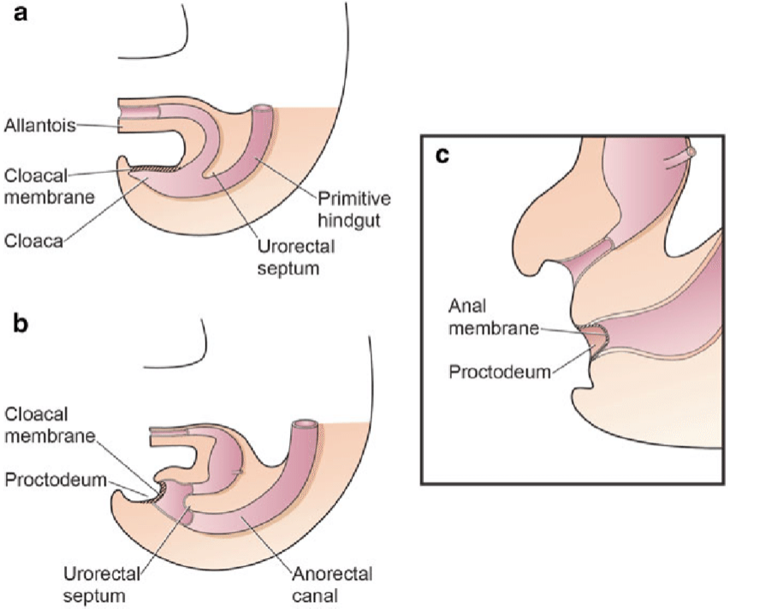
Figura 1 División de la cloaca en un seno urogenital primitivo anterior y un recto posterior entre las semanas 4 y 6 de gestación
La teoría más relacionada del desarrollo embrionario en la extrofia, sostenida por Marshall y Muecke,6 describe el defecto básico como un sobredesarrollo anómalo de la porción inferior de la membrana cloacal, que impide la migración medial del tejido mesenquimatoso. Por lo tanto, no se produce el desarrollo adecuado de la pared abdominal. El momento de la ruptura de este defecto cloacal determina la gravedad del trastorno. Las perforaciones centrales que dan lugar a la extrofia clásica tienen la mayor incidencia (60%), mientras que las variantes de extrofia representan el 30% y la extrofia cloacal el 10%.
Se han propuesto otras teorías sobre la causa del complejo extrofia-epispadias.7,8 Ambrose y O’Brian9 postularon que un desarrollo anormal de los tubérculos genitales, con fusión en la línea media por debajo y no por encima de la membrana cloacal, produce el defecto de extrofia. Otra hipótesis describe una inserción caudal anómala del tallo corporal con falta de interposición del tejido mesenquimatoso en la línea media.10 Debido a esta falla, no se produce el desplazamiento de la cloaca hacia las porciones profundas de la cavidad abdominal. Una membrana cloacal que permanece en una posición infraumbilical superficial representa un estado embrionario inestable con una fuerte tendencia a desintegrarse.11 Ninguna teoría parece dilucidar todos los aspectos del complejo observado clínicamente y continúan los estudios para describir por completo el proceso de desarrollo que finalmente forma el complejo extrofia-epispadias.
Etiología
La etiología exacta de esta enfermedad aún no ha sido identificada. No obstante, se ha confirmado que el evento desencadenante ocurre en etapas tempranas del embarazo.8 Múltiples estudios en curso sugieren una posible base genética para el desarrollo de estas afecciones.11 Existe una mayor incidencia en hijos de madres que recibieron dosis altas de progesterona en etapas tempranas del embarazo, como en casos de terapias de reproducción asistida, y se estima que la incidencia es hasta 7,5 veces mayor en los casos en los que se utilizó la fertilización in vitro.11
Riesgo de recurrencia
El posible papel de los factores genéticos implicados en la expresión de la EEC se sustenta en el mayor riesgo de recurrencia para la descendencia de individuos afectados.8 Entre hermanos, Ives et al estimaron que el riesgo de recurrencia era de aproximadamente el 1% cuando los padres de los casos de BEX no eran consanguíneos ni estaban afectados.12 Shapiro et al.13 estimaron el riesgo de recurrencia para los hermanos de origen europeo en la BEX aislada como 1 en 275, tras haber encuestado a 2.500 familias con CEB. También describieron un riesgo 400 veces mayor de BEX clásica en la descendencia de individuos afectados en comparación con la población general. Con base en una prevalencia de 3,3:100.000 (~1:30.000) de BEX aislada entre poblaciones de origen europeo, se ha calculado que la razón del riesgo de recurrencia en hermanos es de 108 (1:275/1:30.000 ~108).14 Si bien las pacientes con EEC representan la minoría de los pacientes, es interesante que solo las mujeres afectadas produjeron descendencia afectada.13 Entre otras explicaciones, este mayor riesgo de recurrencia aparente en mujeres afectadas podría deberse a una mayor predisposición genética a la EEC (el llamado efecto de Carter).15
Recurrencias familiares
Aunque la agregación familiar es rara, 30 familias multiplex reportadas respaldan la idea de una susceptibilidad genética subyacente a la EEC.16 En la mayoría de estas familias, dos miembros están afectados. Sin embargo, en casos infrecuentes la herencia de la EEC puede ser consistente con una herencia autosómica dominante con penetrancia reducida o con un rasgo autosómico recesivo o con transmisión ligada al cromosoma X.17 Estas observaciones indican que existen uno o más genes con un efecto importante sobre el fenotipo, aunque en la mayoría de los casos son necesarios factores causales adicionales para que el fenotipo se manifieste. Un pequeño subgrupo de casos puede seguir una herencia mendeliana, mientras que en la mayoría, la EEC se hereda como un rasgo complejo con múltiples factores genéticos (heredables o mutaciones *de novo *somáticas o de la línea germinal), y complejas interacciones gen-gen, o gen-ambiente que contribuyen a su formación.8
Genética molecular del EEC
Los análisis citogenéticos y moleculares han revelado anomalías cromosómicas en 20 pacientes con EEC hasta la fecha, aunque ninguna de ellas parece ser causal.8 Se observaron aberraciones cromosómicas numéricas en seis pacientes. En otros cuatro varones con BEX, una mujer con BEX y una niña con CEX, se encontró una asociación con el síndrome de Down.16 La aneuploidía de los cromosomas sexuales en cinco de estos casos podría señalar a un locus gonosómico implicado en la formación de la EEC. Se han identificado aberraciones estructurales en seis casos de EEC y en un paciente que presentaba simultáneamente CEX e hipomelanosis de Ito. Aunque los puntos de ruptura exactos no se han determinado en ninguno de estos casos, se detectaron varias translocaciones que involucraban la región q32-ter del cromosoma 9.
Agentes teratogénicos y EEC
Los estudios de gemelos y los datos epidemiológicos sugieren que los factores ambientales desempeñan un papel en la etiología de la EEC. Sin embargo, los estudios epidemiológicos existentes no han identificado factores teratogénicos mayores.4,12,18 Varios estudios han confirmado que el sexo masculino, la raza, la edad parental avanzada4 y la mayor paridad, incluso tras ajustar por la edad,19 son factores de riesgo predisponentes. Gambhir et al.18 describieron que la exposición materna periconcepcional al tabaquismo era significativamente más frecuente en pacientes con CEX que en un grupo combinado de pacientes con epispadias y BEX clásica. Varios informes describieron la aparición de EEC en lactantes resultantes de *in vitro *fertilización, pero sigue siendo objeto de debate si la incidencia de niños con EEC concebidos mediante *in vitro *fertilización parece ser mayor de lo esperado.8,20
Presentación clínica
Periodo prenatal
A pesar de la magnitud del defecto en la pared abdominal inferior y en el desarrollo de los órganos pélvicos, la extrofia vesical sigue siendo difícil de diagnosticar de manera fiable mediante ecografía prenatal.21 Es probable que esto se deba a su baja incidencia y a que a menudo se confunde con diagnósticos más frecuentes como onfalocele o gastrosquisis.
Es posible sospechar estas afecciones durante el embarazo cuando:
- La vejiga no se identifica en ecografías sucesivas (ausencia de llenado vesical).
- Hay una disminución del grosor de la pared abdominal.
- Hay una masa en el abdomen inferior que se vuelve más prominente a medida que avanza el embarazo.
- Hay un cordón umbilical de implantación baja.
- Los genitales tienen una posición anómala (anterior o posterior) y hay dificultades para determinar el sexo del bebé
- El falo es corto.
- Hay un aumento del diámetro pélvico, con separación de las ramas púbicas.
- Hay un onfalocele.
- Hay evidencia de anomalías de las extremidades inferiores y/o mielomeningocele (sugestivo de extrofia cloacal).
La ecografía tridimensional y el uso creciente de la resonancia magnética fetal mejorarán la capacidad para diagnosticar la extrofia vesical y cloacal.21 El diagnóstico prenatal permite el asesoramiento prenatal y realizar los arreglos para el parto en un centro especializado en extrofia. Esto permite un enfoque multidisciplinario por equipos con experiencia en el manejo de la naturaleza única del complejo extrofia-epispadias.
Período neonatal
La mayoría de las variantes son fácilmente identificables al nacer.
- La extrofia vesical se observa más frecuentemente en recién nacidos a término con buen peso al nacer.
- Los recién nacidos con extrofia cloacal suelen ser prematuros y pequeños para la edad gestacional.
Infancia
- Las variantes poco frecuentes pueden pasar desapercibidas.
- Por lo general se identifican más tarde en la vida solo por incontinencia urinaria persistente o alteraciones de la marcha.
Examen físico
En el BEX clásico, la mayoría de las anomalías consisten en defectos de la pared abdominal, la vejiga, los genitales, los huesos pélvicos y el ano, implicando así el tracto urinario inferior, los genitales y el sistema musculoesquelético (extremidades), mientras que en la extrofia cloacal existe una mayor afectación del tracto gastrointestinal y del SNC.
Extrofia vesical clásica
La pared abdominal está alargada, y el ombligo está de implantación baja, ubicado en el borde superior de la placa vesical; puede asociarse con un defecto herniario o un pequeño onfalocele. La vejiga está abierta anteriormente, con su mucosa completamente expuesta; pueden observarse pólipos en la superficie. La orina drena desde los orificios ureterales en la superficie vesical. El cierre tardío puede conducir a alteraciones inflamatorias o mecánicas adicionales con signos de inflamación de la mucosa como un recubrimiento blanquecino, ulceraciones y formaciones hiperplásicas. Las bandas de piel fina y brillante alrededor de la extrofia marcan la transición entre la piel normal y el área de metaplasia escamosa. El ano está más anterior, pero la función esfinteriana es normal. Los huesos púbicos están ampliamente separados y también pueden estar acortados y en rotación externa (30%). Son palpables a ambos lados de la plantilla vesical y del extremo distal de los bordes triangulares. En la mayoría de los pacientes de ambos sexos se palpan hernias inguinales bilaterales.
- BEX femenino: hay un clítoris bífido, labios mayores separados y un mons pubis divergente (Figura 2). La vagina es más corta de lo normal, de no más de 6 cm de profundidad, pero de calibre normal. El orificio vaginal suele estar estenótico y ubicado anteriormente. Como el ano también está posicionado ventralmente, el periné está acortado.
- BEX masculino: El defecto genital es grave y probablemente sea uno de los aspectos más problemáticos de la reconstrucción quirúrgica. La placa uretral está abierta y se extiende a lo largo de un falo corto, ancho y curvado dorsalmente, desde la vejiga abierta hasta el surco glandular. Ambos cuerpos cavernosos se encuentran por debajo de la placa uretral. Un examen cuidadoso revela el colículo seminal y los conductos eyaculadores como diminutos orificios en la zona, donde presumiblemente la próstata se ubica dorsalmente. El glande está abierto y plano (Figura 3), y los testículos de tamaño normal suelen estar ubicados en el escroto.
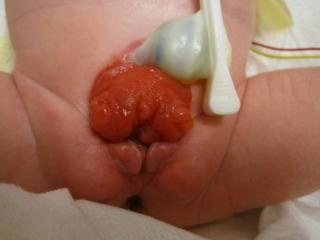
Figura 2 BEX clásica con ombligo de implantación baja y ano situado anteriormente en el periné. BEX femenina con vejiga abierta y placa uretral, con clítoris bífido.
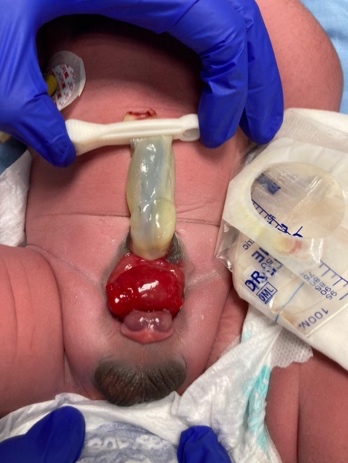
Figura 3 BEX masculino con un falo corto, ancho y curvado dorsalmente. Distancia anormal entre el escroto y el pene.
Epispadias
El defecto resulta de una detención del desarrollo en términos de falta de cierre de la placa uretral y, adicionalmente, de una localización uretral dorsal anormal. Por lo tanto, en varones se encuentra un meato ectópico o una tira de mucosa en el dorso del pene y en mujeres se detecta una hendidura uretral de grado variable.8 La pared abdominal y los músculos rectos del abdomen, así como el ombligo, están completamente desarrollados de manera normal. La sínfisis púbica suele estar cerrada, o solo hay una diástasis pequeña, lo que indica únicamente anomalías leves de la pelvis y del suelo pélvico. La incontinencia urinaria parece ser el principal síntoma clínico, en función del grado de afectación del esfínter urinario. En la mayoría de los epispadias distales no se observa pérdida involuntaria de orina, mientras que en los casos proximales la orina gotea de manera permanente a través del meato. Debido a las a veces “leves” anomalías clínicas, el epispadias distal puede pasarse por alto al nacimiento, especialmente en niñas. Entonces, el diagnóstico puede realizarse más tarde, en edad escolar, debido a una incontinencia urinaria resistente al tratamiento estándar.
- **Epispadias masculino (Figura 4): Según la localización del meato, el epispadias se distingue en penopúbico, peniano o glandular.
- **Epispadias femenino (Figura 5): se divide en tres grados según Davis, ya sea leve con un meato patuloso, intermedio o grave con una hendidura que compromete toda la uretra y el cuello vesical, que además presenta prolapso de mucosa vesical.
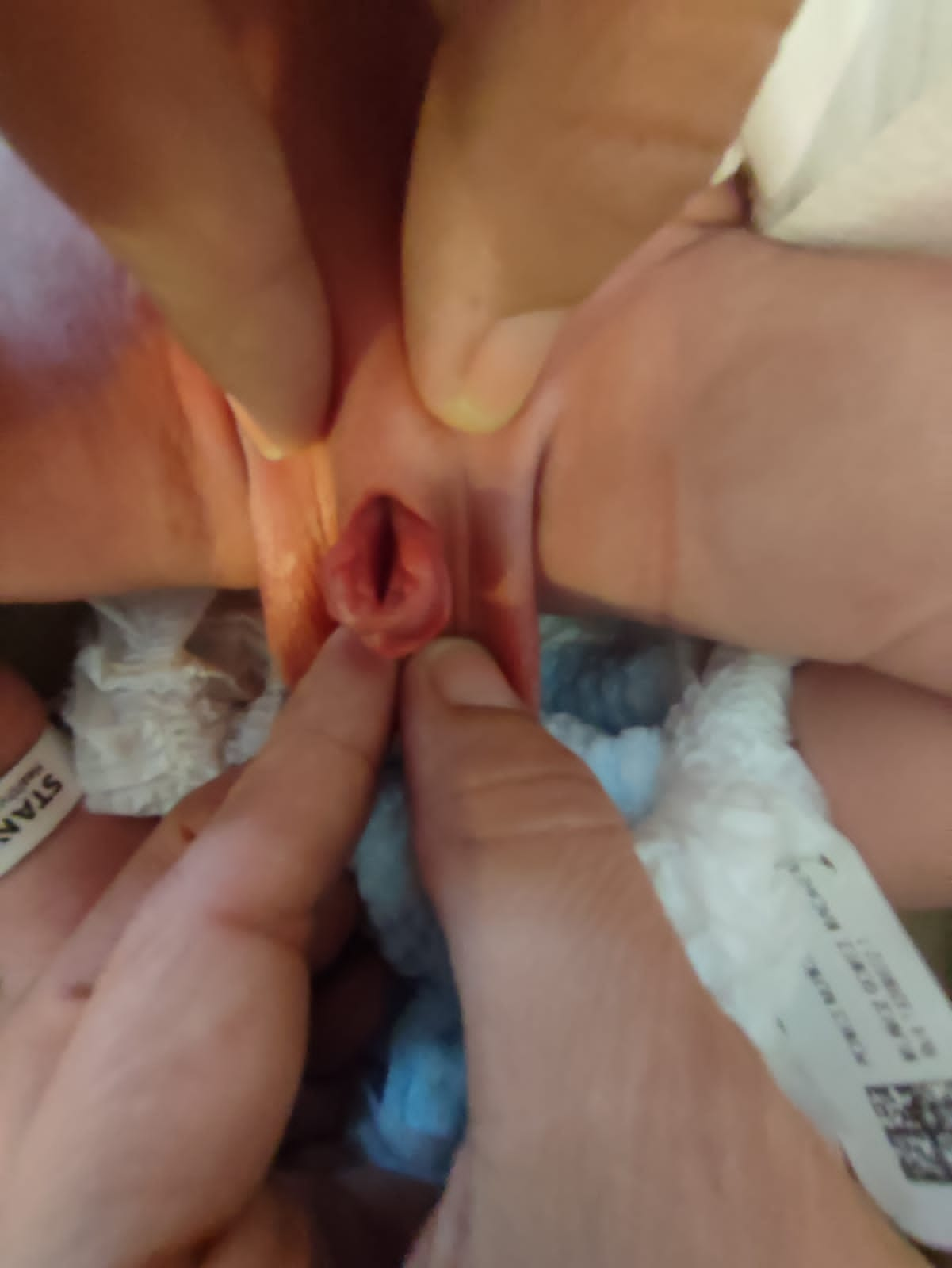
Figura 4 Epispadias masculino
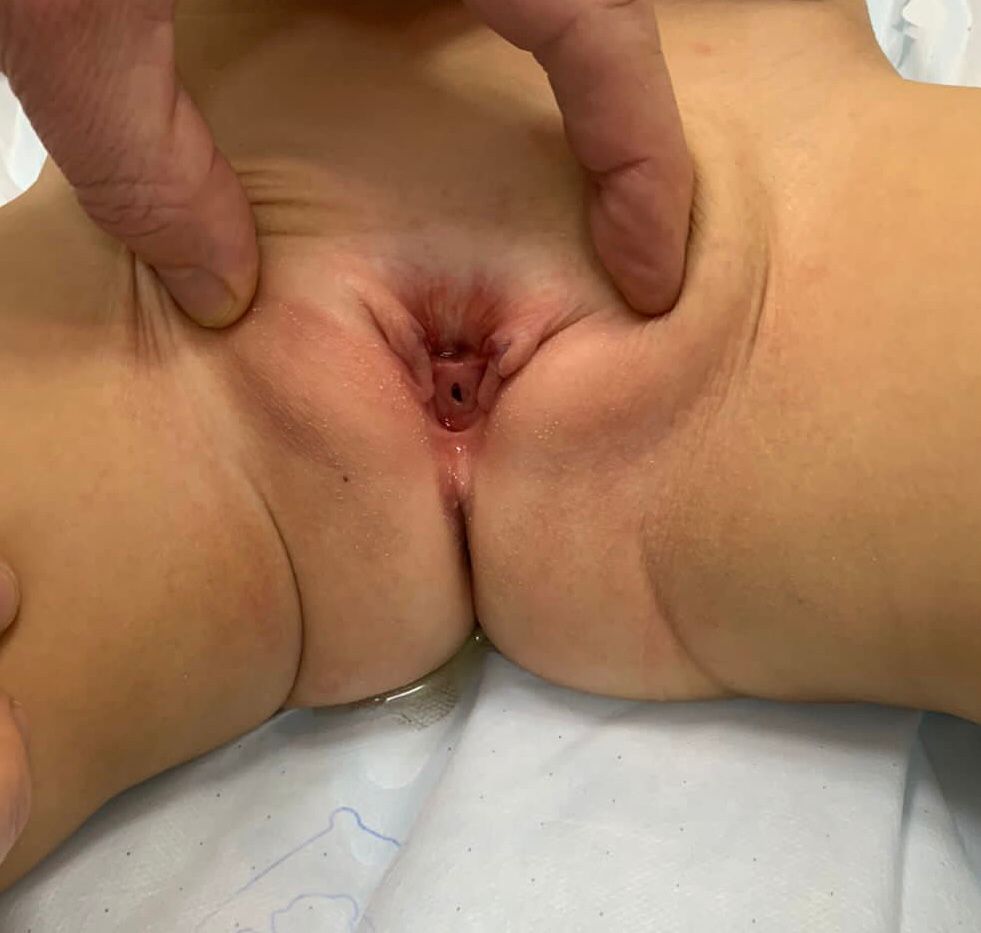
Figura 5 Epispadias femenino
Extrofia cloacal
Los músculos rectos y los huesos púbicos están separados. La vejiga está abierta en la pared abdominal inferior y dividida en 2 mitades adyacentes al segmento expuesto del ciego. Los orificios que comunican con el íleon terminal, el apéndice (uno o dos) y el intestino distal son evidentes dentro de la placa cecal, y el íleon terminal puede prolapsar a través de ella como una “trompa” (apariencia de trompa de elefante). Se presenta con ano imperforado y puede asociarse a un onfalocele. El 95% presenta mielodisplasia y el 65% presenta una malformación de las extremidades inferiores.
-
CEX masculino: el falo suele ser bífido y pequeño, con cada hemiglande situado caudal a cada hemivejiga, o puede estar ausente.
-
CEX femenina: el clítoris es bífido y puede haber dos hemivaginas con un útero bicorne (Figura 6).
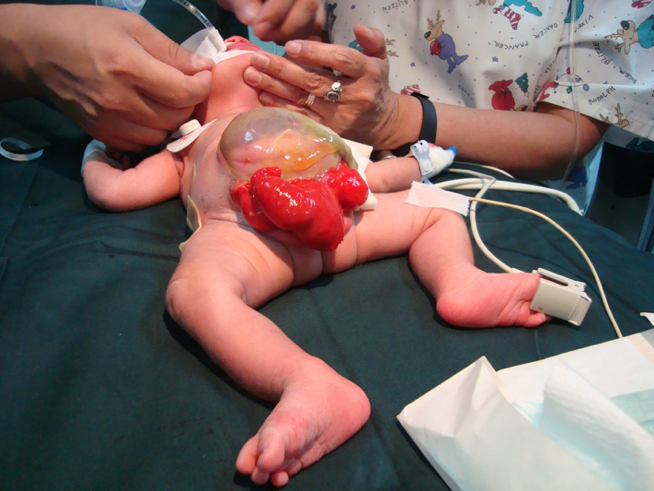
Figura 6 Extrofia cloacal. Onfalocele gigante con 2 hemivejigas adyacentes a la placa cecal extrofiada. Falo pequeño y bífido, con su hemiglande y un hemiescroto situados distalmente a cada placa vesical.
Variantes de la extrofia
Esto incluye un espectro clínicamente amplio y heterogéneo de anomalías. La extrofia cubierta se parece a la BEX clásica, con parte de la mucosa vesical cubierta por piel; también puede presentarse como un epispadias grave con una vejiga prolapsada. El ombligo puede estar en una posición ortotópica. La pseudoextrofia, sin embargo, puede ser muy difícil de identificar después del nacimiento y con frecuencia se detecta en edades más avanzadas. Los genitales parecen normales, y los pacientes pueden no presentar ningún síntoma urinario; algunos son completamente continentes. En la exploración física puede encontrarse diástasis de los rectos de diversos grados. Las radiografías pélvicas podrían demostrar una sínfisis púbica abierta, aunque no es infrecuente que sea un hallazgo incidental.
Anomalías asociadas
Anomalías urológicas
Varias malformaciones urológicas están presentes en aproximadamente un tercio de todos los casos de EEC, predominantemente en la población con EC (obstrucción de la unión pieloureteral, riñón pélvico ectópico, riñón en herradura, agenesia renal, megauréter, ectopia ureteral y ureterocele).1,8 Todos los pacientes deben someterse a un procedimiento antirreflujo con cada plastia del cuello vesical, ya que existe una prevalencia del 100% de reflujo vesicoureteral bilateral debido a un defecto del desarrollo de la unión ureterovesical en todo el espectro de la EEC.
Anomalías espinales y ortopédicas
La incidencia de anomalías espinales varía dentro del espectro EEC. En los niños nacidos con CEB, las anomalías espinales se presentan en aproximadamente el 7% de los casos, mientras que un grupo heterogéneo de anomalías espinales congénitas resultantes del cierre defectuoso del tubo neural en etapas tempranas de la vida fetal y del desarrollo anómalo de la masa celular caudal puede confirmarse mediante resonancia magnética (RM) y se ha identificado en casi el 100% de los pacientes con CEX.8 Debe tenerse en cuenta un componente neurológico en estos pacientes con respecto a la función vesical y de las extremidades inferiores, y la capacidad eréctil.22,23 Las anomalías esqueléticas y de las extremidades (deformidades de pie equinovaro, ausencia de pies, deformidades tibiales o peroneas y luxaciones de cadera) se observan con mayor frecuencia en CEX.1,8 Sin embargo, no hay informes sobre displasia de cadera en el seguimiento a largo plazo del EEC.
Anomalías gastrointestinales
Estas se asocian predominantemente con CEX. Además de un remanente común del intestino posterior de tamaño variable, se encuentran onfaloceles en el 88-100% de los casos de CEX. La malrotación o duplicación gastrointestinal, así como el síndrome de intestino corto, pueden observarse en hasta el 46% de los casos.1,8 En aproximadamente el 25% de los casos, un síndrome de intestino corto, ya sea anatómico o funcional, provoca malabsorción.
Anomalías ginecológicas
Además de las anomalías de los genitales externos, el cuello uterino se inserta muy bajo en la pared vaginal superior, cerca del introito en la mayoría de los casos.1,8,24 No obstante, la anatomía y la función del útero y de los anexos son normales. El suelo pélvico y el defecto del elevador del ano, junto con la ausencia de los ligamentos cardinales, predisponen a las mujeres al prolapso vaginal o uterino en alrededor del 50% de los casos. Las anomalías müllerianas son bastante frecuentes en la CEX, es decir, duplicación de la vagina y del útero, agenesia vaginal.1,8
Estudios de imagen
Suelen realizarse para identificar y diagnosticar posibles anomalías asociadas (malformaciones pélvicas y abdominales, anomalías de la columna vertebral y de la médula espinal, etc).
Los estudios principales son la ecografía renal, la ecografía medular y las radiografías de la columna vertebral y la pelvis. Todos los recién nacidos con CEX deben someterse a ecografía medular y radiografías para definir las anomalías espinales individuales, que van desde hemivértebra hasta mielomeningocele. Además, se recomienda realizar resonancia magnética en el seguimiento para identificar anomalías espinales ocultas que predisponen a un anclaje medular sintomático.
La evaluación ecográfica de las articulaciones de la cadera es de importancia fundamental para todos los pacientes con EEC. La radiografía pélvica simple puede ser útil para estimar la dimensión de la diástasis de la sínfisis y la localización de las caderas.
Diagnóstico
En el periodo prenatal, se puede sospechar el EEC a partir de la semana 16 de gestación. Con los avances en la sensibilidad de la ecografía antenatal, ha habido un aumento en la sospecha prenatal.
En la mayoría de los casos, la malformación se diagnostica al nacer, ya que se hace evidente clínicamente.
Manejo
Si existe un diagnóstico prenatal o sospecha de EEC, lo ideal es asesorar a los padres y el parto debe programarse en un centro especializado (remitir si es necesario). No existe experiencia que avale la indicación de cirugía fetal. El parto vaginal no se recomienda debido al mayor riesgo de lesión de la placa vesical.
En la actualidad, ya no se considera una emergencia y/o emergencia quirúrgica, lo que permite un traslado en condiciones seguras a centros especializados que pueden atender a estos niños con malformaciones de baja frecuencia y alta complejidad.
El enfoque quirúrgico se centró inicialmente en la derivación urinaria para preservar la función renal. Sin embargo, desde que Young informó del primer cierre vesical exitoso en 1942,1,8,11,24,25 se han logrado avances significativos en las áreas de la reconstrucción por etapas, en la apariencia de los genitales externos y en la preservación de la continencia y la función renal. Con mejoras en la técnica quirúrgica, así como en la atención perioperatoria, ahora se puede esperar una excelente supervivencia a largo plazo.
Cirugía
Los objetivos del enfoque quirúrgico actual son;1 reconstrucción de la pared abdominal,2 recolocación y cierre anatómico de la vejiga extrofiada,3 la preservación de la función renal y la consecución de la continencia urinaria, y4 reconstrucción de los genitales externos.26,27,28,29,30 Históricamente, la reconstrucción se realizaba en 3 etapas comenzando en el período neonatal. Sin embargo, en la actualidad existen centros superespecializados o equipos multicéntricos que manejan la afección y pueden organizar un manejo diferido, con el fin de ofrecer lo mejor a cada paciente mediante la acumulación de experiencia en patologías de baja frecuencia y alta complejidad.31,32,33,34,35,36,37,38,39,40
Las etapas quirúrgicas son
- Cierre de la vejiga, con aproximación de los huesos púbicos, sin necesidad de osteotomías. En pacientes con CEX, se debe realizar una derivación intestinal.
- La segunda etapa consiste en la reconstrucción genital completa. En algunos centros, también se realiza el Procedimiento de Kelly o una Reconstrucción Radical de Tejidos Blandos, para lograr la continencia vesical y una mayor longitud peneana en los varones.
- Finalmente, la continencia urinaria debe evaluarse a mediano y largo plazo. Para aquellos casos en los que aún no se ha alcanzado este objetivo o cuando existe riesgo de compromiso de la función renal, hay una tercera etapa en la que se puede realizar cirugía del cuello vesical y/o cistoplastia de aumento con una derivación urinaria continente para cateterismo intermitente limpio.
En diversos centros superespecializados de todo el mundo, se ha optado por el cierre diferido a los dos a cuatro meses de edad, realizando un abordaje en un solo tiempo, junto con la cirugía descrita por Kelly,26,30,39 que incluye:1 disección y cierre vesical con o sin un procedimiento antirreflujo (si el tamaño vesical y las características de la pared lo permiten);2 la movilización radical de tejidos blandos con desinserción de las ramas púbicas;3 la creación de un cuello vesical o de un mecanismo de continencia con ayuda del estimulador de Peña para identificar el complejo esfinteriano neomuscular y4 reconstrucción peneana o introitoplastia.
Los beneficios de este enfoque son que se evita una cirugía previa en el período neonatal, permitiendo la preservación de los tejidos nativos y evitando la cicatrización y la fibrosis generadas por el cierre primario. Esto también favorece el apego parental y el desarrollo de un vínculo entre el niño y sus padres, sin necesidad de una hospitalización prolongada durante los primeros días de vida.
Se ha demostrado que el cierre vesical diferido puede realizarse de forma segura y con éxito, y que en principio no afecta el desarrollo de la vejiga. Posponer la cirugía permite la maduración del recién nacido, con la consiguiente reducción de los riesgos de una anestesia prolongada y favorece el crecimiento del lactante durante el período de mini-pubertad, además de promover la preparación del equipo de expertos para cada caso.39
Complicaciones
Debido a su complejidad y muy baja frecuencia, estas afecciones presentan una serie de complicaciones principalmente asociadas a la cirugía reconstructiva:34,39
- Precoces: dehiscencia de herida quirúrgica, prolapso vesical, fístulas uretrales o vesicocutáneas (4 – 19%), estenosis uretral (8%), isquemia peneana.
- Tardías: RVU, infecciones urinarias recurrentes, vaciamiento vesical incompleto, incontinencia urinaria, litiasis vesical, fibrosis de la pared abdominal, ruptura vesical, prolapso uterino, insuficiencia renal, eyaculación retrógrada, oligozoospermia e infertilidad, y síndrome de intestino corto e incontinencia fecal (extrofia cloacal).
Pronóstico
Si estos pacientes no reciben tratamiento, sufrirían infecciones del tracto urinario recurrentes, incontinencia urinaria permanente y problemas con la actividad sexual, además de presentar un mayor riesgo de cáncer de vejiga, lo que en conjunto supone una peor calidad de vida.26,27,28,29,30,31,32,33,34,35
Sin embargo, si se realiza un manejo adecuado de la afección tal como se sugiere, estos pacientes tienen una expectativa de calidad de vida que probablemente sea similar a la de la población general. También se estima que potencialmente podrán llevar una vida absolutamente normal, considerando que en más del 90% de los casos no existe asociación con otras malformaciones congénitas. Además, estos pacientes podrían desarrollar continencia urinaria sin requerir un aumento vesical o cateterismo intermitente para el vaciamiento, aunque la tasa de éxito es reservada (solo el 23% de micción voluntaria por uretra con la técnica MSRE,41 39% con la técnica CPRE,28 y 53% con la técnica de Kelly26).
Tabla 1 Amplio rango de las tasas de continencia de los distintos enfoques según la definición de continencia y el período de observación.42
| Enfoque | Tasa de continencia (%) | Literatura |
|---|---|---|
| MSRE | 74 | Gearhart et al |
| 62 | Gupta et al | |
| 22 | Dickson et al | |
| CPRE | 80 | Grady et al |
| 74 | Hammouda et al | |
| 23 | Arab et al | |
| RSTM | 73 | Kelly et al |
| 70 | Jarzebowski et al | |
| 33–67 (femenino) | Cuckow et al | |
| 44–81 (masculino) |
Actualmente, estos pacientes tienen una excelente supervivencia a largo plazo, logrando continencia urinaria en hasta un 75-80% de los pacientes con extrofia vesical y en un 65-70% en los casos de extrofia cloacal;39,43,41 sin embargo, muchos de ellos requieren derivaciones urinarias e intestinales permanentes. En general, la función sexual se preserva y la mayoría de los pacientes son fértiles. Se recomienda el parto por cesárea en las pacientes sometidas a cirugía para evitar daño a los mecanismos de continencia.
Referencias
- Gearhart JP, Gearhart JP, Rink RC. The Bladder Exstrophy–epispadias–cloacal Exstrophy Complex. J Pediatr Urol 2001: 386–415. DOI: 10.1016/b978-1-4160-3204-5.00030-x.
- Siffel C, Correa A, Amar E, Bakker MK, Bermejo-Sánchez E, Bianca S, et al.. Bladder exstrophy: An epidemiologic study from the International Clearinghouse for Birth Defects Surveillance and Research, and an overview of the literature. Am J Med Genet C Semin Med Genet 2011; 157 (4): 321–332. DOI: 10.1002/ajmg.c.30316.
- Cervellione RM, Mantovani A, Gearhart J, Bogaert G, Gobet R, Caione P, et al.. Prospective study on the incidence of bladder/cloacal exstrophy and epispadias in Europe. J Pediatr Urol 2015; 11 (6): 337.e1–337.e6. DOI: 10.1016/j.jpurol.2015.03.023.
- Boyadjiev SA, Dodson JL, Radford CL, Ashrafi GH, Beaty TH, Mathews RI, et al.. Clinical and molecular characterization of the bladder exstrophy-epispadias complex: analysis of 232 families. BJU Int. 2004;94:1337-1343. . DOI: 10.1111/j.1464-410X.2004.05170.x..
- Männer J, Kluth D. The morphogenesis of the exstrophy-epispadias complex: a new concept based on observations made in early embryonic cases of cloacal exstrophy. Anat Embryol (Berl) 2005; 210 (1): 51–57. DOI: 10.1007/s00429-005-0008-6.
- Marshall VF, Muecke EC. Variations in exstrophy of the bladder. Plast Reconstr Surg 1962; 31 (4): 396. DOI: 10.1097/00006534-196304000-00030.
- Stephens FD, Hutson JM. Differences in embryogenesis of epispadias, exstrophy–epispadias complex and hypospadias. J Pediatr Urol 2005; 1 (4): 283–288. DOI: 10.1016/j.jpurol.2005.01.008.
- Ebert AK, Reutter H, Ludwig M, Rösch WH. Exstrophy-epispadias complex. Definitions 2009; 4 (23). DOI: 10.32388/7eqi40.
- Ambrose SS, O’Brien DP. Surgical Embryology of the Exstrophy-Epispadias Complex. Surg Clin North Am 1974; 54 (6): 1379–1390. DOI: 10.1016/s0039-6109(16)40493-7.
- Mildenberger H, Kluth D, Dziuba M. Embryology of bladder exstrophy. J Pediatr Surg. 1988;23:166-170. . DOI: 10.1016/s0022-3468(88)80150-7..
- Gearhart JP. Exstrophy-Epispadias Complex. Campbell-Walsh-Wein Urology, vol. 31. 12th ed. 2021. DOI: 10.1016/b978-1-4160-6911-9.00124-9.
- Ives E, Coffey R, Carter CO. A family study of bladder exstrophy. J Med Genet. 1980;17:139-141. . DOI: 10.1136/jmg.17.2.139..
- Shapiro E, Lepor H, Jeffs RD. The inheritance of the exstrophy-epispadias complex. J Urol. 1984;132:308-310. .
- Reutter H, Qi L, Gearhart JP, Boemers T, Ebert AK, Rösch WH, et al.. Concordance analyses of twins with bladder exstrophy-epispadias complex suggest genetic etiology. Am J Med Genet A. 2007;143:2751-2756. . DOI: 10.1002/ajmg.a.31975.
- Carter CO. Genetics of common single malformations. Br Med Bull. 1976;32:21-26. .
- Ludwig M, Ching B, Reutter H, Boyadjiev SA. The bladder exstrophy-epispadias complex. Birth Defects Res Part A Clin Mol Teratol. 2009;85:509-22. . DOI: 10.1002/bdra.20557..
- Reutter H, Shapiro E, Gruen JR. Seven new cases of familial isolated bladder exstrophy and epispadias complex (BEEC) and review of the literature. Am J Med Genet A. 2003;120A:215-221. . DOI: 10.1002/ajmg.a.20057..
- Gambhir L, Höller T, Müller M, Schott G, Vogt H, Detlefsen B, et al.. Epidemiological survey of 214 European families with Bladder Exstrophy-Epispadias Complex (BEEC) J Urol. 2008;179:1539-1543. . DOI: 10.1016/j.juro.2007.11.092..
- Byron-Scott R, Haan E, Chan A, Bower C, Scott H, Clark K. A population-based study of abdominal wall defects in South Australia and Western Australia. Paediatr Perinat Epidemiol. 1998;12:136-151. . DOI: 10.1046/j.1365-3016.1998.00090.x..
- Wood HM, Babineau D, Gearhart JP. In vitro fertilization and the cloacal/bladder exstrophy-epispadias complex: A continuing association. J Pediatr Urol. 2007;3:305-310. . DOI: 10.1016/j.jpurol.2006.10.007..
- Palmer B, Frimberger D, Kropp B. Bladder exstrophy-epispadias complex and cloacal exstrophy. Section 7.D: Developmental anomalies. Paediatric Urology Book. Paediatric Urology Department, University of Oklahoma; 2011. DOI: 10.1007/978-1-84882-132-3\\_45.
- Rösch WH, Hanisch E, Hagemann M, Neuhuber WL. The Characteristic innervation pattern of the urinary bladder in particular forms of exstrophy-epispadias-complex. BJU. 2001;87:30. .
- Schober JM, Carmichael PA, Hines M, Ransley PG. The ultimate challenge of cloacal exstrophy. J Urol. 2002;167:300-304. . DOI: 10.1016/s0022-5347(05)65455-9..
- Woodhouse CRJ, Hinsch R. The anatomy and reconstruction of the adult female genitalia in classical exstrophy. BJU. 1997;79:618-622. . DOI: 10.1046/j.1464-410X.1997.00148.x..
- Woodhouse. C.R.J.: Genitoplasty in exstrophy and epispadias. Cambridge University Press; 2006, DOI: 10.1017/cbo9780511545757.046.
- Kelly JH. Vesical exstrophy: repair using radical mobilisation of soft tissues. Pediatr Surg Int 1995; 10 (5-6): 298–304. DOI: 10.1007/bf00182207.
- Grady RW, Mitchell ME. Complete primary repair of exstrophy. J Urol. 1999; 62 (4): 415–1420. DOI: 10.1097/00005392-199910000-00071.
- Groth. Bladder exstrophy consortium (MIBEC) after 5 years. AUA Chicago; 2019.
- Dickson AP. The management of bladder exstrophy: the Manchester experience. J Pediatr Surg. 2014; 9 (2): 44–250. DOI: 10.1016/j.jpedsurg.2013.11.031.
- Cuckow P, López PJ. Bladder Exstrophy Closure and Epispadias. In: Spitz L, Coran A, editors. En Operative Pediatric Surgery. 7th ed. 2013. DOI: 10.1201/b13237-101.
- Borer JG, Gargollo PC, Hendren WH, Diamond DA, Peters CA, Atala A, et al.. Early Outcome Following Complete Primary Repair Of Bladder Exstrophy In The Newborn. J Urol 2005; 174 (4 Part 2): 1674–1679. DOI: 10.1097/01.ju.0000175942.27201.59.
- Baird AD, Nelson CP, Gearhart JP. Modern staged repair of bladder exstrophy: a contemporary series. J Pediatr Urol 2007. 4: 11–315. DOI: 10.1016/j.jpurol.2006.09.009.
- Borer JG, Vasquez E, Canning DA, Kryger JV, Mitchell ME. An initial report of a novel multi-institutional bladder exstrophy consortium: a collaboration focused on primary surgery and subsequent care. J Urol. 2015. DOI: 10.1016/j.juro.2014.10.114.
- Ellison JS, Shnorhavorian M, Willihnganz-Lawson K, Grady R, Merguerian PA. A critical appraisal of continence in bladder exstrophy: Long-term outcomes of the complete primary repair. J Pediatr Urol 2016. 2 (4): 05 1–205 2057. DOI: 10.1016/j.jpurol.2016.04.005.
- Schaeffer AJ, Stec AA, Purves JT, Cervellione RM, Nelson CP, Gearhart JP. Complete primary repair of bladder exstrophy: a single institution referral experience. J Urol. 2011; 86 (3): 041–1046. DOI: 10.1016/j.juro.2011.04.099.
- Pathak P, Ring JD, Delfino KR, Dynda DI, Mathews RI. Complete primary repair of bladder exstrophy: a systematic review. J Pediatr Urol. 2020; 6 (2): 49–153. DOI: 10.1016/j.jpurol.2020.01.004.
- Ahn JJ, Shnorhavorian M, Katz C, Goldin AB, Merguerian PA. Early versus delayed closure of bladder exstrophy: A National Surgical Quality Improvement Program Pediatric analysis. J Pediatr Urol. 2018; 4 (1): 7 1–27 5. DOI: 10.1016/j.jpurol.2017.11.008.
- Leclair MD, Villemagne T, Faraj S, Suply E. The radical soft-tissue mobilization (Kelly repair) for bladder exstrophy. J Pediatr Urol. 2015; 1 (6): 64–365. DOI: 10.1016/j.jpurol.2015.08.007.
- Leclair MD, Faraj S, Sultan S. One-stage combined delayed bladder closure with Kelly radical soft-tissue mobilization in bladder exstrophy: preliminary results. J Pediatr Urol. 2018; 4 (6): 58–564. DOI: 10.1016/j.jpurol.2018.07.013.
- Baradaran N, Stec AA, Schaeffer AJ, Gearhart JP, Mathews RI. Delayed primary closure of bladder exstrophy: immediate postoperative management leading to successful outcomes. Urology. 2012; 9 (2): 15–419. DOI: 10.1016/j.urology.2011.08.077.
- Jarzebowski AC, McMullin ND, Grover S SR, BR H, J.M.. The Kelly technique of bladder exstrophy repair: continence, cosmesis and pelvic organ prolapse outcomes. J Urol. 2009. DOI: 10.1016/j.juro.2009.02.083.
- Cervellione RM, Husmann DA, Bivalacqua TJ, Sponseller PD, Gearhart JP. Penile ischemic injury in the exstrophy/epispadias spectrum: new insights and possible mechanisms. J Pediatr Urol. 2010; 5: 50–456. DOI: 10.1016/j.jpurol.2010.04.007.
- Purves JT, Gearhart JP. Complications of radical soft-tissue mobilization procedure as a primary closure of exstrophy. J Pediatr Urol 2008. 1: 5–69. DOI: 10.1016/j.jpurol.2007.02.006.
- Maruf M, Manyevitch R, Michaud J. Urinary Continence Outcomes in Classic Bladder Exstrophy: A Long-Term Perspective. J Urol. 2020; 03 (1): 00–205. DOI: 10.1097/ju.0000000000000505.
- Promm M, Roesch WH. Recent Trends in the Management of Bladder Exstrophy: The Gordian Knot Has Not Yet Been Cut. Front Pediatr 2019; 7. DOI: 10.3389/fped.2019.00110.
Última actualización: 2025-09-25 12:10