
12: Anomalias Congênitas do Rim e do Trato Urinário
Este capítulo levará aproximadamente 14 minutos para ler.
Introdução
Anomalias congênitas dos rins e do trato urinário (CAKUT) são um grupo de anomalias estruturais que afetam os rins e o trato urinário. Estima-se que existam aproximadamente 4 a 60 casos por 10.000 nascimentos na população geral. O diagnóstico de CAKUT é tipicamente feito durante um exame de ultrassonografia pré-natal de rotina ou no período pós-natal em um recém-nascido com sinais e sintomas clínicos associados.1 A prevalência de CAKUT em recém-nascidos prematuros foi de 2% em uma grande coorte recente, reportada, com 409704 lactentes.2
A anomalia mais comum é a obstrução da junção ureteropélvica, que afeta aproximadamente 20% dos indivíduos com a condição. Entre os outros tipos de CAKUT, há rins displásicos multicísticos, agênese renal, displasia renal, hipoplasia renal, refluxo vesicoureteral, megaureter, sistema coletor duplo, ureter ectópico e válvulas uretrais posteriores (Figura 1).
Apesar dos avanços significativos na detecção e no diagnóstico da CAKUT, as causas subjacentes da condição ainda não são totalmente compreendidas. São necessárias mais pesquisas para aprimorar nossa compreensão dos fatores embriológicos, genéticos e ambientais que contribuem para o desenvolvimento de CAKUT e para compreender melhor suas causas subjacentes e melhorar as estratégias de prevenção e tratamento.
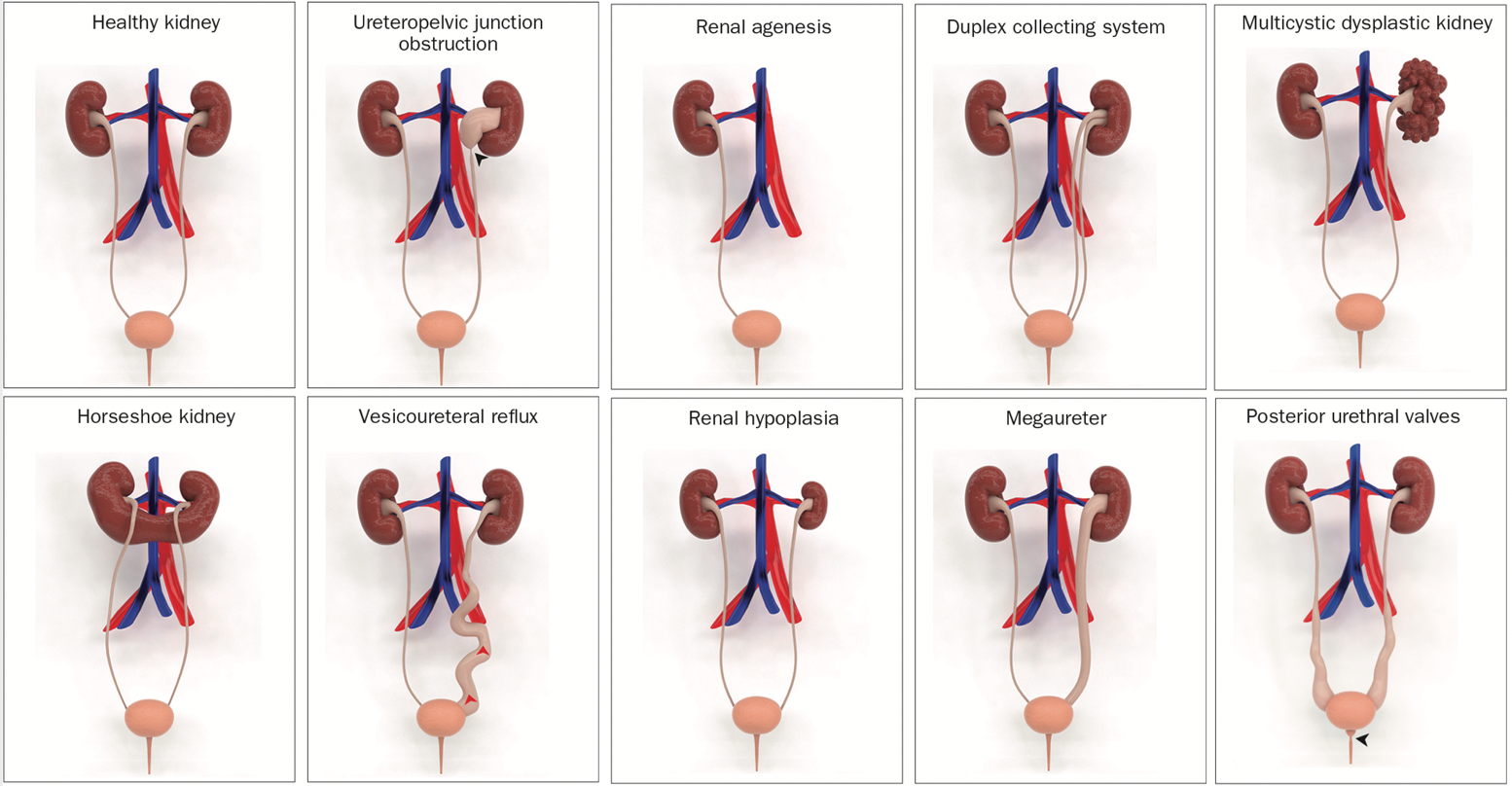
Figura 1 Modelos 3D ilustrativos de anomalias congênitas do rim e do trato urinário (CAKUT).3
Embriologia
O desenvolvimento dos rins e do trato urinário no feto envolve um processo complexo que se inicia nas primeiras semanas de gestação e envolve a formação e a diferenciação de várias estruturas. Esse processo começa com a formação dos brotos ureterais, que surgem do mesoderma intermediário e dão origem aos ductos coletores e, por fim, aos rins. Os rins também se desenvolvem a partir de uma estrutura chamada metanéfros, que se forma a partir do mesoderma no embrião.
Durante o desenvolvimento, os brotos ureterais e o metanefro devem conectar-se e diferenciar-se adequadamente para formar os rins funcionais e o trato urinário. Interrupções nesse processo podem levar a uma variedade de anomalias congênitas.
CAKUT pode ser categorizada por localização (rins, ureteres, bexiga ou uretra), por causa (mutações genéticas, exposições ambientais, infecções durante a gravidez etc.), por herança e por gravidade (Tabela 1). Em 10% a 25% dos casos, CAKUT é atribuída a doenças genéticas. CAKUT exibe manifestações fenotípicas complexas em associação com doenças genéticas (Figura 2 e Figura 3).4 É possível que indivíduos com as mesmas anormalidades do genoma se apresentem com diferentes formas de CAKUT, e anomalias fenotípicas semelhantes podem surgir devido a diferentes doenças genéticas e estar associadas a doenças extrarrenais.2
Tabela 1 Categorias dos distúrbios CAKUT
| Rim | Sistema coletor |
|---|---|
| Número: Agenesia renal | Obstrução da junção ureteropélvica |
| Morfologia: Hipoplasia renal, displasia, MCDK | Refluxo vesicoureteral |
| Posição: Em ferradura, ectópico, rins pélvicos | Megaureter |
| Sistema duplo | |
| PUV |
Várias malformações do parênquima renal resultam na falha do desenvolvimento normal dos néfrons, incluindo displasia cística, displasia renal, agenesia renal, disgenesia tubular renal e disgenesia tubular renal.
Doença renal policística
O termo doença renal policística é geralmente usado para descrever uma doença na qual há múltiplos cistos nos rins sem displasia associada.
A doença renal policística autossômica dominante (ADPKD) e a doença renal policística autossômica recessiva (ARPKD) são os dois tipos mais comuns de doença renal policística. Além do padrão de herança, seja ele dominante ou recessivo, ARPKD e ADPKD diferem quanto às suas manifestações clínicas e prognósticos. A ADPKD é a forma mais comum de PKD e é causada por mutações nos genes PKD1 ou PKD2.
Genes implicados no desenvolvimento da doença renal cística:5,6
- PKD1: Este gene está associado à ADPKD. Mutações no gene PKD1 são responsáveis por cerca de 85% dos casos de ADPKD.
- PKD2: Este gene também está associado à ADPKD. Mutações no gene PKD2 são responsáveis por cerca de 15% dos casos de ADPKD.
- HNF1β: Mutações neste gene estão associadas a uma doença renal policística autossómica dominante tipo 2 (ADPKD2).
- REN: Este gene está associado a uma doença renal policística infantil.
- MUC1: Mutações neste gene estão associadas a uma doença renal cística medular relacionada ao MUC1.
- INVS: Mutações neste gene estão associadas a uma doença renal cística medular relacionada com a nefronoftise.
- NPHP1, NPHP3, NPHP4, NPHP5, NPHP6, NPHP7, NPHP8, NPHP9: Estes genes estão associados a uma doença renal cística medular relacionada com a nefronoftise.
- BMP7: Mutações neste gene estão associadas a uma doença renal cística medular relacionada com a nefronoftise
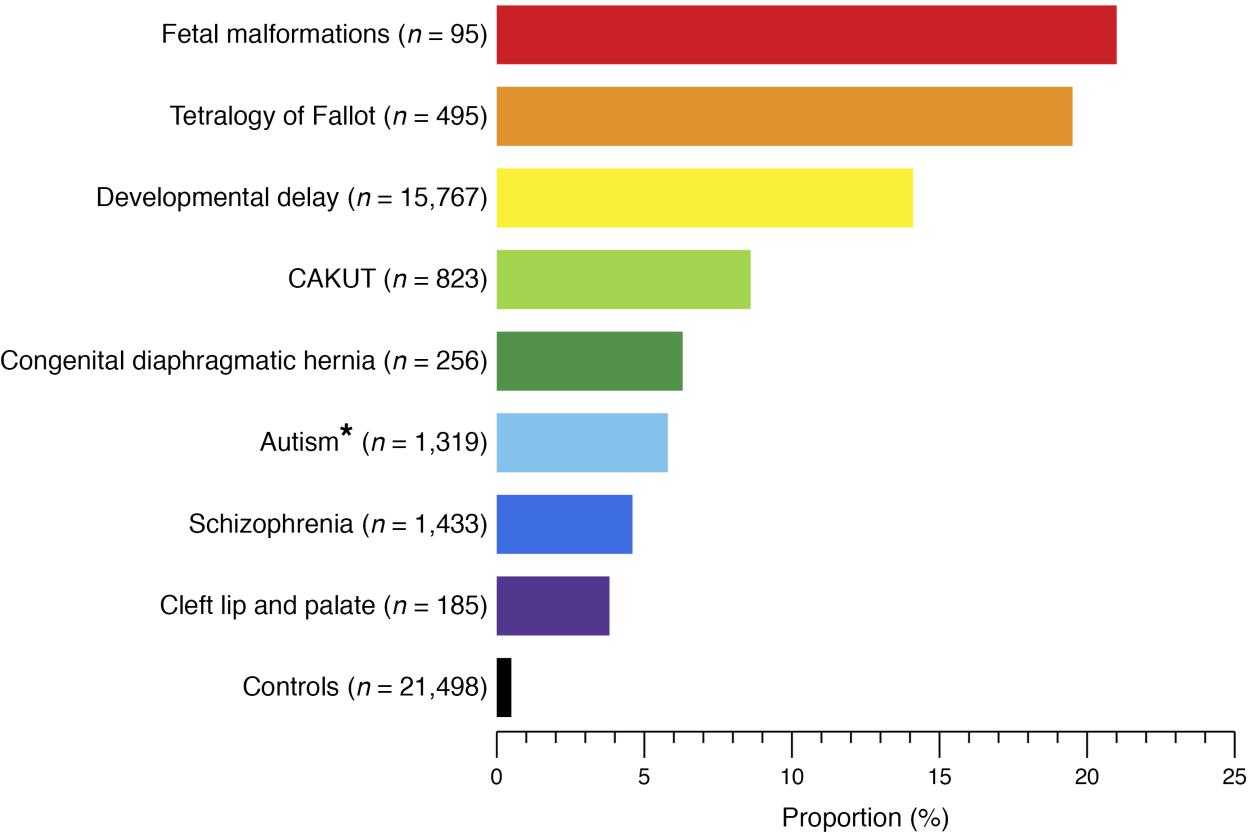
Figura 2 Proporção de pacientes com distúrbios genômicos conhecidos em diferentes fenótipos do desenvolvimento humano e controles saudáveis.4
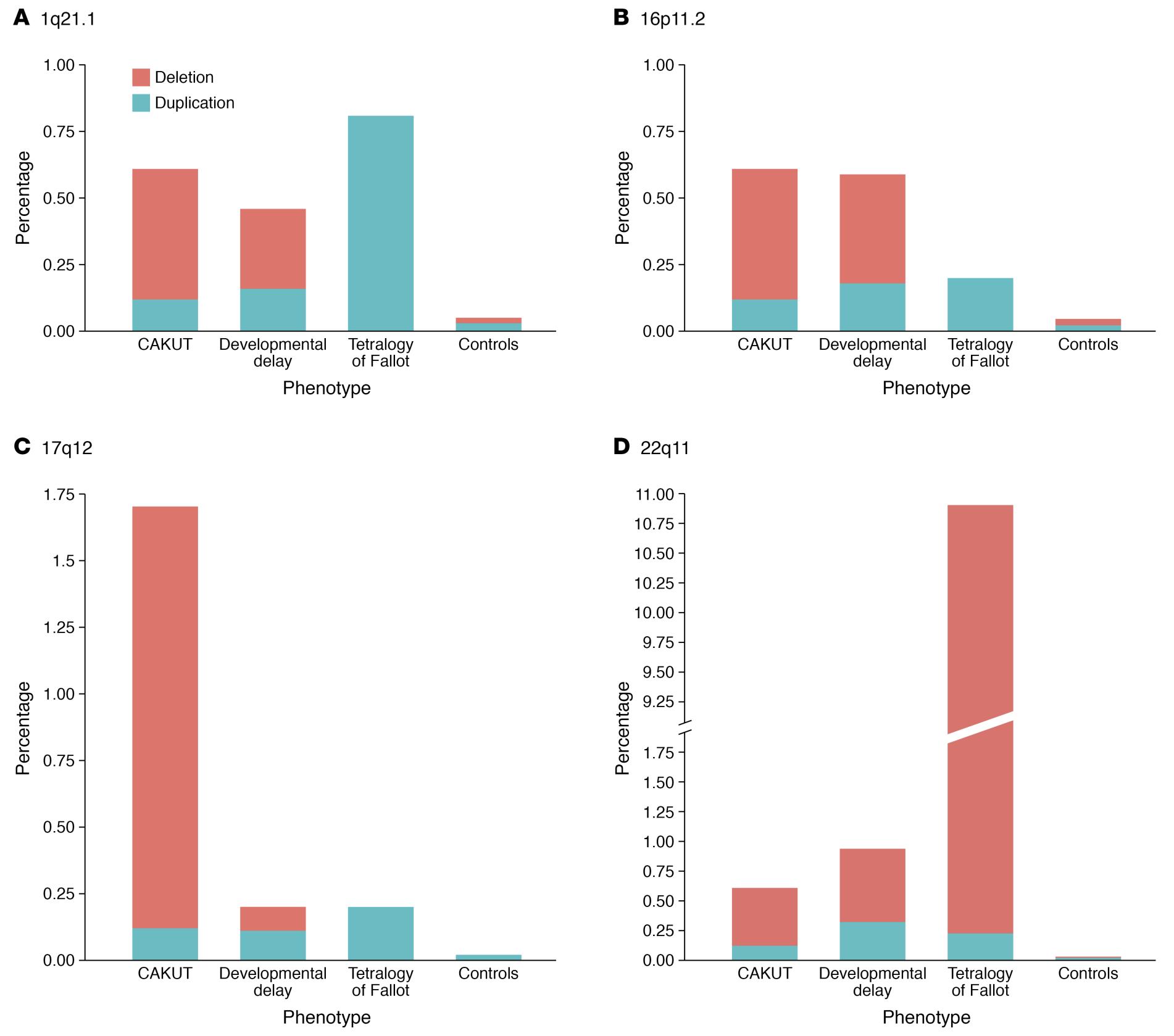
Figura 3 Diferenças e semelhanças na prevalência dos quatro lóci de CNV mais comumente implicados em pacientes com CAKUT.4
MCDK
MCDK é uma doença cística congênita não hereditária, um tipo de displasia renal que causa múltiplos cistos não comunicantes separados por parênquima displásico. A prevalência de MCDK é de aproximadamente 1 em 3.600 a 1 em 4.300 nascidos vivos.7,8 A causa de MCDK é desconhecida, mas acredita-se que possa estar relacionada à formação anormal do broto ureteral, à exposição a teratógenos ou à obstrução do trato urinário. MCDK é frequentemente detectada por meio de ultrassonografia pré-natal. Como frequentemente não causa sintomas ou complicações, geralmente só é identificada no período pós-natal se houver uma massa perceptível ou se for descoberta incidentalmente durante exames de imagem para outra condição.
Alguns dos genes que têm sido associados à MCDK incluem:9,10,11
- HNF1β: Este gene codifica uma proteína chamada fator nuclear dos hepatócitos 1 beta, que desempenha um papel no desenvolvimento e na função do rim e de outros órgãos. Mutações em HNF1β podem causar uma variedade de anomalias renais, incluindo MCDK.
- PAX2: Este gene codifica uma proteína chamada caixa pareada 2, que é importante para o desenvolvimento do ureter, o tubo que transporta a urina do rim para a bexiga. Mutações em PAX2 podem causar MCDK e outras anomalias renais.
- SIX1: Este gene codifica uma proteína chamada sine oculis homeobox 1, que está envolvida no desenvolvimento de vários órgãos, incluindo o rim. Mutações em SIX1 podem causar MCDK e outras anomalias renais.
- WT1: Este gene codifica uma proteína chamada tumor de Wilms 1, que é importante para o desenvolvimento do rim e de outros órgãos. Mutações em WT1 podem causar MCDK e outras anomalias renais.
O MCDK é tipicamente manejado de forma conservadora (isto é, com observação), uma vez que a maioria dos casos não apresenta complicações a longo prazo. Pacientes com anomalias contralaterais podem apresentar comprometimento renal ao longo do tempo. Assim, é necessário acompanhamento a longo prazo.
Agenesia Renal
Quando comparada à agenesia renal bilateral, a agenesia renal unilateral ocorre em uma a cada 1.000 gestações e apresenta prognóstico favorável. Estima-se que entre 0,1% e 0,3% de 1.000 nascimentos sejam afetados por agenesia renal bilateral, o que geralmente resulta em taxas de sobrevivência baixas. Vários padrões de herança foram descritos em famílias com agenesia renal, que pode ocorrer como um achado isolado ou como parte de uma síndrome.12
A agenesia renal unilateral pode ser causada por mutações em vários genes, incluindo ATRX, RET, BMP4, FRAS1, FREM1, GRIP1, GLI3, UPK3A, entre outros. Além disso, a agenesia renal bilateral pode ser causada por mutações nos genes RET, FGF20, ITGA8. Genes relacionados listados na Tabela 2.13,14
Displasia Renal
A hipodisplasia renal resulta de uma interrupção do desenvolvimento durante o primeiro trimestre da vida fetal do blastema renal metanéfrico. Entretanto, a causa da hipodisplasia renal permanece desconhecida; anomalias vasculares in utero, assim como distúrbios genéticos do desenvolvimento, são dois mecanismos possíveis. É possível que a hipodisplasia renal seja um achado isolado ou parte de uma síndrome genética associada a malformações do trato urinário. Uma análise de genes do desenvolvimento (HNF1β, PAX2, EYA1, SIX1 e SALL1) envolvidos na displasia renal, em uma grande coorte de crianças com hipodisplasia renal, foi fornecida pelo Estudo ESCAPE. De acordo com esse estudo, mutações em PAX2 foram diagnosticadas em 15% dos pacientes com patologia renal, e aqueles com deleções genéticas ou mutações em genes extensos apresentaram fenótipos renais variáveis. Além disso, 22% de todos os pacientes com hipodisplasia renal cística apresentavam uma mutação em HNF1, sugerindo que indivíduos com displasia renal cística sejam rastreados para mutações em HNF1.15 Achados de ultrassonografia renal que mostram redução do tamanho renal e perda da diferenciação corticomedular são clinicamente indicativos de hipodisplasia renal. A evolução para doença renal terminal pode ocorrer em alguns pacientes com hipodisplasia renal ao longo do tempo. Não há tratamento específico para a hipodisplasia renal. É importante acompanhar e manejar a função renal ao longo dos anos. Para os pacientes que evoluem para doença renal terminal, o transplante renal é a opção preferencial.
Junção ureteropélvica
A causa patológica mais comum de hidronefrose neonatal é a obstrução da parte superior do ureter. A incidência global é de 1:1.500, com uma proporção de 2:1 em recém-nascidos do sexo masculino em relação ao feminino.16 Uma obstrução da junção ureteropélvica (JUP) é geralmente causada por estenose intrínseca ou, menos frequentemente, por um vaso cruzado que provoca compressão extrínseca na parte superior do ureter. Um possível mecanismo é a ocorrência de uma disrupção embrionária no ureter proximal que altera o desenvolvimento da musculatura circular, modificando as fibras colágenas e a composição entre e ao redor das células musculares.
Várias mutações monogênicas estão implicadas na UPJO, incluindo Id2, PAX2, EYA, AGTR2, BMP4, SOX17, CHD1L e DSTYK.17,18 Há relatos de que Id2 e Adamts1 produzem um fenótipo semelhante ao fenótipo humano de UPJO. Camundongos desprovidos de Id2 também apresentam inserção alta do ureter na pelve renal.19
Refluxo vesicoureteral
Vários genes foram identificados como associados ao desenvolvimento de VUR. Alguns desses genes estão envolvidos no desenvolvimento e na função do músculo liso ureteral. Outros genes estão envolvidos no desenvolvimento e na função da bexiga. A elevada prevalência de VUR entre gêmeos, irmãos e descendentes sugere uma base genética para essa doença. Está estabelecido que VUR é uma doença geneticamente heterogênea com base nos resultados de estudos genéticos em várias populações. Um estudo de ligação e associação em todo o genoma associou VUR primário não sindrômico à região 10q26 do genoma.20,21 Outros genes que foram associados ao desenvolvimento de VUR incluem o gene FOXC1, o gene MYH11 e o gene ACTN4. Esses genes desempenham vários papéis no desenvolvimento e na função do trato urinário.22,23 Um estudo de associação em todo o genoma utilizando a maior análise de variantes no número de cópias e estudo de associação em todo o genoma de VUR está resultando na identificação de cinco lócus que podem estar associados a VUR, incluindo WDPCP, OTX1, BMP5, WDPCP e WNT5 com grandes efeitos. WNT5 pode desempenhar um papel no desenvolvimento do sistema urogenital. Um estudo de associação em todo o genoma utilizando a maior análise de variantes no número de cópias e estudo de associação em todo o genoma de VUR está resultando na identificação de cinco lócus que podem estar associados a VUR, incluindo WDPCP, OTX1, BMP5, WDPCP e WNT5 com grandes efeitos. WNT5 pode desempenhar um papel no desenvolvimento do sistema urogenital.24
Válvulas uretrais posteriores
A válvula uretral posterior é uma anomalia congênita do trato urinário potencialmente fatal que é geralmente descoberta durante o período neonatal e, apesar do tratamento ideal, resulta em altas taxas de insuficiência renal. Estima-se que PUV ocorra em 1 em 7,000–8,000 nascidos vivos.25
Há uma ampla variedade de relatos que indicam que a PUV é a principal causa de doença renal crônica em recém-nascidos do sexo masculino, e ESRD pode ocorrer numa faixa de 5% a 64% dos casos.26,27 Uma revisão sistemática relatou que a doença renal crônica (CKD) e a doença renal em estágio terminal (ESKD) estão associadas a pacientes com PUV com risco de até 32% e 20%, respectivamente.28
Até o momento, poucos genes foram identificados como associados às válvulas uretrais posteriores. A síndrome de Prune Belly foi relacionada a HNF1B em apenas 3% dos casos reconhecidos, e a maioria dos diagnosticados não apresenta causa genética. Entre as anomalias do CAKUT associadas à trissomia 21, incluem-se válvulas uretrais posteriores, pieliectasia e megaureteres.29,30
Tabela 2 Um resumo dos genes envolvidos no desenvolvimento de CAKUT.31,32,33,22,23,34,35,36
| Anomalia | Genes |
|---|---|
| Hipoplasia/Displasia renal | ACE, AGT, ATRX, CHD7, ESCO2, EYA1, SIX1, FRAS1, FREM2, GATA3, GLI3, GPC3, GRIP1, HNF1β, JAG1, KAL1, LRP4, MKS1–4, NIPBL, PAX2, PBX1, REN, SALL1, SIX5 |
| Agenesia renal | ATRX, BMP4, CBP/EP300, CHD7, ESCO2, FGF20, FRAS1, FREM2, GATA3, GRIP1, GLI3, HNF1β, ITGA8, JAG1, KAL1, KAL2, RET, SALL1, VANGL1 |
| Displasia cística | CBP/EP300, DHCR7, EVC, EVC2, FRAS1, FREM2, GPC-3, HNF1β, JAG1, KAL1, KAL2, MKS1–4, NS1, PEX, PAX2, WT1 |
| Sistema duplicado | NS1 |
| Rim em ferradura/Rim ectópico | GLI3, NIPBL, VANGL1 |
| Hidronefrose, JUP | ACE, Adamts-1, AGT, ATRX, BMP4, CHD1L, CHD7, DHCR7, DSTYK, ESCO2, EYA, FRAS1, FREM2, GLI3, HSPG2, JAG1, Id2, NS1, PAX2, PEX, RET, SOX17, VANGL1 |
| Hidroureter, Megaureter | CHD7, GLI3 |
| RVU | ATRX, DHCR7, EYA1, FOX1, GATA3, HOXA13, HPSE2, JAG1, KAL1, KAL2, MYH11, NIPBL, PAX2, SALL1, SIX1, SIX5 |
| VUP | BNC2, SALL1 |
Importância clínica e implicações futuras do conhecimento genético
À medida que o campo da avaliação genética continua a avançar, é provável que vejamos uma capacidade crescente de identificar as causas genéticas de CAKUT e de prever a probabilidade de desenvolver essas condições. Isso pode levar ao diagnóstico e ao tratamento mais precoces, o que pode melhorar os desfechos para os indivíduos afetados.
Deve-se notar que o teste genético nem sempre é necessário para diagnosticar CAKUT. Um diagnóstico definitivo geralmente é feito com base em uma combinação de apresentação clínica, histórico médico e estudos de imagem. O teste genético para CAKUT geralmente é realizado em um centro especializado sob a supervisão de um geneticista clínico, pois a interpretação desses resultados pode ser complexa e ter implicações para decisões reprodutivas. Vários laboratórios (PreventionGenetics, Invitae, Blueprint Genetics, etc.) oferecem testes genéticos para diferentes distúrbios de CAKUT. O painel de testes para CAKUT desses laboratórios inclui testes genéticos para múltiplos genes associados a distúrbios de CAKUT, incluindo HNF1B, NPHP1 e UPK1B. Os testes genéticos para esses distúrbios podem ajudar a confirmar um diagnóstico, fornecer informações sobre a gravidade do distúrbio e auxiliar no aconselhamento genético e no planejamento familiar. Vale ressaltar que outras empresas e laboratórios de testes genéticos também podem ter um painel para CAKUT. Como CAKUT é um grupo de distúrbios causados por diferentes mutações genéticas, o teste de painel genético para CAKUT em algumas empresas pode não incluir todas as causas genéticas de CAKUT, e diferentes laboratórios podem ter listas diferentes de genes cobertos.
A compreensão completa dos componentes genéticos de CAKUT é essencial para desenvolver estratégias precisas de testes genéticos. Além disso, essa compreensão auxiliará na orientação da tomada de decisões clínicas em relação aos testes genéticos. Incluindo desfechos clínicos significativos e função renal, são necessários estudos longitudinais para investigar os efeitos a longo prazo do diagnóstico e do manejo precoces de CAKUT.
Conclusão e Resumo
As anomalias congênitas do rim e do trato urinário (CAKUT) são anormalidades estruturais que afetam os rins e o trato urinário. Estima-se que existam cerca de 4–60 casos por 10.000 nascimentos. O diagnóstico de CAKUT é geralmente feito durante uma ultrassonografia pré-natal ou após o nascimento em recém-nascidos com sinais e sintomas clínicos associados. A anomalia mais comum é a obstrução da junção ureteropélvica, que afeta aproximadamente 20% das pessoas com a condição. O desenvolvimento dos rins e do trato urinário envolve a formação e diferenciação de várias estruturas, incluindo os brotos ureterais e os metanefros, que devem se conectar adequadamente para formar os rins e o trato urinário funcionais. Alterações nesse processo podem levar à CAKUT. Existem várias categorias de distúrbios de CAKUT, incluindo os que afetam o número, a morfologia e a posição dos rins, e os que afetam o sistema coletor. Em 10–25% dos casos, CAKUT é causada por distúrbios genéticos. Vários genes foram identificados que estão associados ao desenvolvimento de diversas doenças relacionadas aos rins. As pesquisas estão em andamento para melhorar nossa compreensão das causas da CAKUT e para desenvolver melhores estratégias de prevenção e tratamento. Apesar de suas limitações e dificuldades atuais, o diagnóstico genético molecular pode oferecer esperança para melhorar o manejo clínico de pacientes com CAKUT no futuro.
Pontos-chave
- As anomalias congênitas dos rins e do trato urinário (CAKUT) são um grupo de anormalidades estruturais que afetam os rins e o trato urinário, com uma estimativa de 4 a 60 casos por 10.000 nascimentos na população geral.
- O desenvolvimento dos rins e do trato urinário no feto envolve um processo complexo que começa nas primeiras semanas da gestação e envolve a formação e diferenciação de várias estruturas. Interrupções nesse processo podem levar a uma variedade de anomalias congênitas.
- A prevalência de CAKUT em recém-nascidos prematuros foi de 2% em uma grande coorte recentemente relatada com 409.704 lactentes.
- A anomalia mais comum é a obstrução da junção ureteropélvica, que afeta aproximadamente 20% dos indivíduos com a condição.
- Em 10-25% dos casos, CAKUT é atribuída a doenças genéticas.
- A avaliação genética está avançando, aumentando a capacidade de identificar as causas genéticas de CAKUT e prever a probabilidade de desenvolver essas condições.
- Os testes genéticos nem sempre são necessários para o diagnóstico de CAKUT; este é tipicamente baseado em uma combinação de apresentação clínica, histórico médico e estudos de imagem.
- Uma compreensão completa dos componentes genéticos de CAKUT é essencial para desenvolver estratégias precisas de testes genéticos e orientar a tomada de decisão clínica na testagem genética.
- São necessários estudos longitudinais para investigar os efeitos a longo prazo do diagnóstico e manejo precoces de CAKUT.
Referências
- Murugapoopathy V, Gupta IR. A Primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin J Am Soc Nephrol 2020; 15 (5): 723–731. DOI: 10.2215/cjn.12581019.
- Hays T, Thompson MV, Bateman DA, Sahni R, Tolia VN, Clark RH, et al.. The Prevalence and Clinical Significance of Congenital Anomalies of the Kidney and Urinary Tract in Preterm Infants. JAMA Netw Open 2022; 5 (9): e2231626. DOI: 10.1001/jamanetworkopen.2022.31626.
- Nicolaou N, Renkema KY, Bongers EMHF, Giles RH, Knoers NVAM. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol 2015; 11 (12): 720–731. DOI: 10.1038/nrneph.2015.140.
- Srivastava S, Molinari E, Raman S, Sayer JA. Many Genes–One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front Pediatr 2018; 5. DOI: 10.3389/fped.2017.00287.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. 8 (1): 25–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 2018; 128 (1): 4–15. DOI: 10.1172/jci95300.
- Gordon AC, Thomas DF, Arthur RJ, Irving HC. Multicystic dysplastic kidney: Is nephrectomy still appropriate? J Pediatr Surg 1988; 24 (9): 951. DOI: 10.1016/s0022-3468(89)80673-6.
- Schreuder MF, Westland R, Wijk JAE van. Unilateral multicystic dysplastic kidney: a meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol Dial Transplant 2009; 24 (6): 1810–1818. DOI: 10.1093/ndt/gfn777.
- Kopač M, Kordič R. Associated Anomalies and Complications of Multicystic Dysplastic Kidney. Pediatr Rep 2022; 14 (3): 375–379. DOI: 10.3390/pediatric14030044.
- Fletcher J, Hu M, Berman Y, Collins F, Grigg J, McIver M, et al.. Multicystic Dysplastic Kidney and Variable Phenotype in a Family with a Novel Deletion Mutation ofPAX2. J Am Soc Nephrol 2005; 16 (9): 2754–2761. DOI: 10.1681/asn.2005030239.
- Chang Y-M, Chen C-C, Lee N-C, Sung J-M, Chou Y-Y, Chiou Y-Y. PAX2 Mutation-Related Renal Hypodysplasia: Review of the Literature and Three Case Reports. Front Pediatr 2022; 9 (765929). DOI: 10.3389/fped.2021.765929.
- Bienstock JL, Birsner ML, Coleman F, Hueppchen NA. Successful In Utero Intervention for Bilateral Renal Agenesis. Obstet Gynecol 2014; 124 (2): 413–415. DOI: 10.1097/aog.0000000000000339.
- Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, et al.. Faculty Opinions recommendation of Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2012; 1 (2): 96–200. DOI: 10.3410/f.13934957.15391059.
- Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal Aplasia in Humans Is Associated with RET Mutations. Am J Hum Genet 2008; 82 (2): 344–351. DOI: 10.1016/j.ajhg.2007.10.008.
- Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al.. Faculty Opinions recommendation of Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2006; 7 (10): 864–870. DOI: 10.3410/f.1047767.508721.
- LEBOWITZ ROBERTL, GRISCOM NTHORNE. Neonatal Hydronephrosis. Radiol Clin North Am 1977; 15 (1): 49–59. DOI: 10.1016/s0033-8389(22)02539-8.
- AC SJ, DM M, oes S, AC S. Congenital Anomalies of the Kidney and Urinary Tract: An Overview. Congenital Anomalies of the Kidney and Urinary Tract 2014; 02 (4): 1–13. DOI: 10.1007/978-3-319-29219-9_1.
- Avanoglu A, Tiryaki S. Embryology and Morphological (Mal)Development of UPJ. Front Pediatr 2020; 8: 137. DOI: 10.3389/fped.2020.00137.
- Aoki Y, Mori S, Kitajima K, Yokoyama O, Kanamaru H, Okada K, et al.. Id2 haploinsufficiency in mice leads to congenital hydronephrosis resembling that in humans. Genes Cells 2004; 9 (12): 1287–1296. DOI: 10.1111/j.1365-2443.2004.00805.x.
- Williams G, Fletcher JT, Alexander SI, Craig JC. Vesicoureteral reflux. Journal of the American Society of Nephrology 2008; 19 (5): 847–862. DOI: 10.1681/asn.2007020245.
- Darlow JM, Darlay R, Dobson MG, Stewart A, Charoen P, Southgate J, et al.. Genome-wide linkage and association study implicates the 10q26 region as a major genetic contributor to primary nonsyndromic vesicoureteric reflux. Sci Rep 2017; 7 (1): 59. DOI: 10.1038/s41598-017-15062-9.
- Woolf AS, Lopes FM, Ranjzad P, Roberts NA. Congenital Disorders of the Human Urinary Tract: Recent Insights From Genetic and Molecular Studies. Front Pediatr 2019; 7 (136). DOI: 10.3389/fped.2019.00136.
- Wu C-HW, Mann N, Nakayama M, Connaughton DM, Dai R, Kolvenbach CM, et al.. Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT). Genet Med 2020; 22 (10): 1673–1681. DOI: 10.1038/s41436-020-0844-z.
- Verbitsky M, Krithivasan P, Batourina E, Khan A, Graham SE, Marasà M, et al.. Review of: "Genome-wide association study followed by trans-ancestry meta-analysis identify 17 new risk loci for schizophrenia". J Am Soc Nephrol 2021; 32 (4): 805–820. DOI: 10.32388/jqb9pr.
- Thakkar D, Deshpande AV, Kennedy SE. Epidemiology and demography of recently diagnosed cases of posterior urethral valves. Pediatr Res 2014; 76 (6): 560–563. DOI: 10.1038/pr.2014.134.
- Warshaw BL, Edelbrock HH, Ettenger RB, Malekzadeh MH, Pennisi AJ, Uittenbogaart CH, et al.. Renal transplantation in children with obstructive uropathy. J Pediatr Surg 1980; 15 (6): 986. DOI: 10.1016/s0022-3468(80)80355-1.
- Roth KS, Carter WH, Chan JCM. Obstructive Nephropathy in Children: Long-Term Progression After Relief of Posterior Urethral Valve. Pediatrics 2001; 107 (5): 1004–1010. DOI: 10.1542/peds.107.5.1004.
- Hennus PML, Kort LMO de, Bosch JLH, Jong TPVM de, Heijden GJMG van der. A Systematic Review on the Accuracy of Diagnostic Procedures for Infravesical Obstruction in Boys. PLoS One 2014; 9 (2): e85474. DOI: 10.1371/journal.pone.0085474.
- Rodriguez MM. Congenital anomalies of the kidney and urinary tract (CAKUT). Lijec Vjesn 2014; 144 (Supp 1). DOI: 10.26800/lv-144-supl1-26.
- Postolache L, Parsa A, Simoni P, Boitsios G, Ismaili K, Schurmans T, et al.. Widespread kidney anomalies in children with Down syndrome. Pediatr Nephrol 2022; 37 (10): 2361–2368. DOI: 10.1007/s00467-022-05455-y.
- Uy N, Reidy K. Developmental Genetics and Congenital Anomalies of the Kidney and Urinary Tract. J Pediatr Genet 2016; 05 (01): 051–060. DOI: 10.1055/s-0035-1558423.
- Kolvenbach CM, Dworschak GC, Frese S, Japp AS, Schuster P, Wenzlitschke N, et al.. Expanding congenital abnormalities of the kidney and urinary tract (CAKUT) genetics: basonuclin 2 (BNC2) and lower urinary tract obstruction. Ann Transl Med 2019; 7 (S6): S226–s226. DOI: 10.21037/atm.2019.08.73.
- Heidet L, Morinière V, Henry C, De Tomasi L, Reilly ML, Humbert C, et al.. Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 2017; 28 (10): 2901–2914. DOI: 10.1681/asn.2017010043.
- Talati AN, Webster CM, Vora NL. Prenatal genetic considerations of congenital anomalies of the kidney and urinary tract (CAKUT). Prenat Diagn 2019; 39 (9): 679–692. DOI: 10.1002/pd.5536.
- Puri P, Gosemann J-H, Darlow J, Barton DE. Genetics of vesicoureteral reflux. Nat Rev Urol 2011; 8 (10): 539–552. DOI: 10.1038/nrurol.2011.113.
- Chen F. Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol 2009; 24 (9): 1621–1632. DOI: 10.1007/s00467-008-1072-y.
Ultima atualização: 2025-09-21 13:35