
12: Congenital Anomalies of the Kidney and Urinary Tract
This chapter will take approximately 14 minutes to read.
Introduction
Congenital anomalies of the kidney and urinary tract (CAKUT) are a group of structural abnormalities that affect the kidneys and urinary tract. It is estimated that there are approximately 4 to 60 cases per 10,000 births in the overall population. A CAKUT diagnosis is typically made during a routine prenatal ultrasound examination or postnatally in a newborn with associated clinical signs and symptoms.1 The prevalence of CAKUT in preterm infants was 2% in recent large, reported cohort with 409704 infants.2
The most common anomaly is ureteropelvic junction obstruction, which affects approximately 20% of individuals with the condition. Among the other types of CAKUT, there is multicystic dysplastic kidneys, renal agenesis, renal dysplasia, renal hypoplasia, vesicoureteral reflux, megaureter, duplex collecting system, ectopic ureter, and posterior urethral valves (Figure 1).
Despite the significant progress made in the detection and diagnosis of CAKUT, the underlying causes of the condition are still not fully understood. Further research is needed to improve our understanding of the embryologic, genetic, and environmental factors that contribute to CAKUT development and to better understand their underlying causes and improve prevention and treatment strategies.
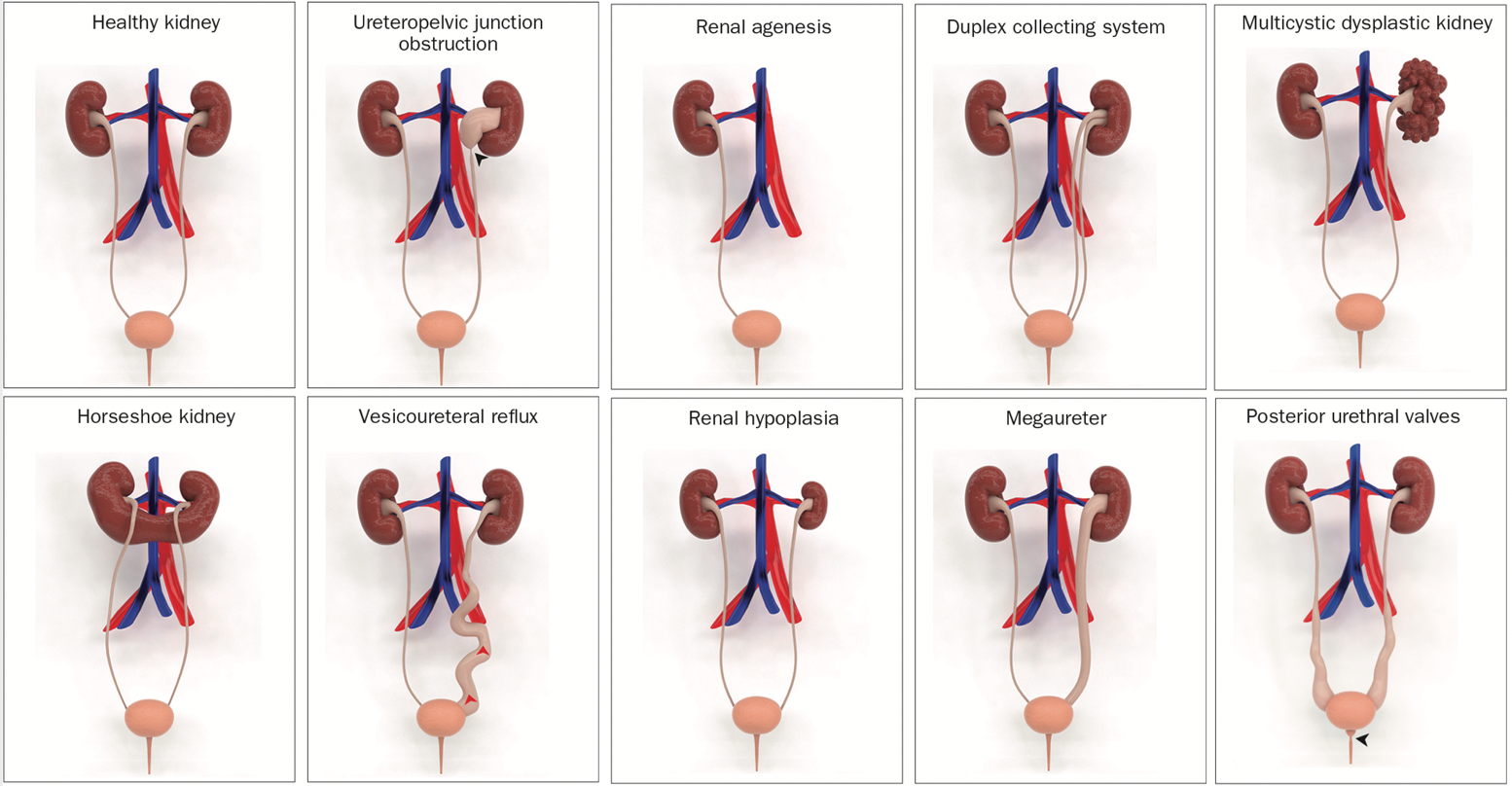
Figure 1 Illustrative 3D models of congenital abnormalities of the kidney and urinary tract (CAKUT).3
Embryology
The development of the kidneys and urinary tract in the fetus involves a complex process that begins in the early weeks of pregnancy and involves the formation and differentiation of several structures. This process begins with the formation of the ureteric buds, which arise from the intermediate mesoderm and give rise to the collecting ducts and ultimately the kidneys. The kidneys also develop from a structure called the metanephros, which forms from the mesoderm in the embryo.
During development, the ureteric buds and metanephros must properly connect and differentiate to form the functional kidneys and urinary tract. Disruptions in this process can lead to a range of congenital anomalies.
CAKUT can be categorized by locations (kidneys, ureters, bladder, or urethra), by cause (genetic mutations, environmental exposures, infections during pregnancy etc.), by inheritance, and by severity (Table 1). In 10% to 25% of cases, CAKUT is attributed to genetic disorders. CAKUT exhibits complex phenotypic manifestations in association with genetic disorders (Figure 2 and Figure 3).4 It is possible for individuals with the same genome abnormalities to manifest with different forms of CAKUT, and similar phenotypic anomalies may arise because of different genetic disorders, and with associated with extrarenal diseases.2
Table 1 Categories of CAKUT disorders
| Kidney | Collecting System |
|---|---|
| Number: Renal agenesis | Ureteropelvic junction obstruction |
| Morphology: Renal hypoplasia, dysplasia, MCDK | Vesicoureteral reflux |
| Position: Horseshoe, ectopic, pelvic kidneys | Megaureter |
| Duplex system | |
| PUV |
Several malformations of the kidney parenchyma result in a failure of the normal development of the nephrons, including cystic dysplasia, renal dysplasia, renal agenesis, renal tubular dysgenesis, and renal tubular dysgenesis.
Polycystic Kidney Disease
The term polycystic kidney disease is generally used to describe a disease in which there are multiple cysts on kidneys without associated dysplasia.
Autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD) are the two most common types of polycystic kidney disease. In addition to the inheritance pattern, whether dominant or recessive inheritance, ARPKD and ADPKD differ in terms of their clinical manifestations and prognoses. ADPKD is the most common form of PKD, and it is caused by mutations in the PKD1 or PKD2 genes.
Genes implicated in the development of renal cystic disease:5,6
- PKD1: This gene is associated with ADPKD. Mutations in the PKD1 gene are responsible for about 85% of cases of ADPKD.
- PKD2: This gene is also associated with ADPKD. Mutations in the PKD2 gene are responsible for about 15% of cases of ADPKD.
- HNF1β: Mutations in this gene are associated with an autosomal dominant polycystic kidney disease type 2 (ADPKD2).
- REN: This gene is associated with an infantile polycystic kidney disease.
- MUC1: Mutations in this gene are associated with a MUC1-related medullary cystic kidney disease.
- INVS: Mutations in this gene are associated with a nephronophthisis-related medullary cystic kidney disease.
- NPHP1, NPHP3, NPHP4, NPHP5, NPHP6, NPHP7, NPHP8, NPHP9: These genes are associated with a nephronophthisis-related medullary cystic kidney disease.
- BMP7: Mutations in this gene are associated with a nephronophthisis-related medullary cystic kidney disease
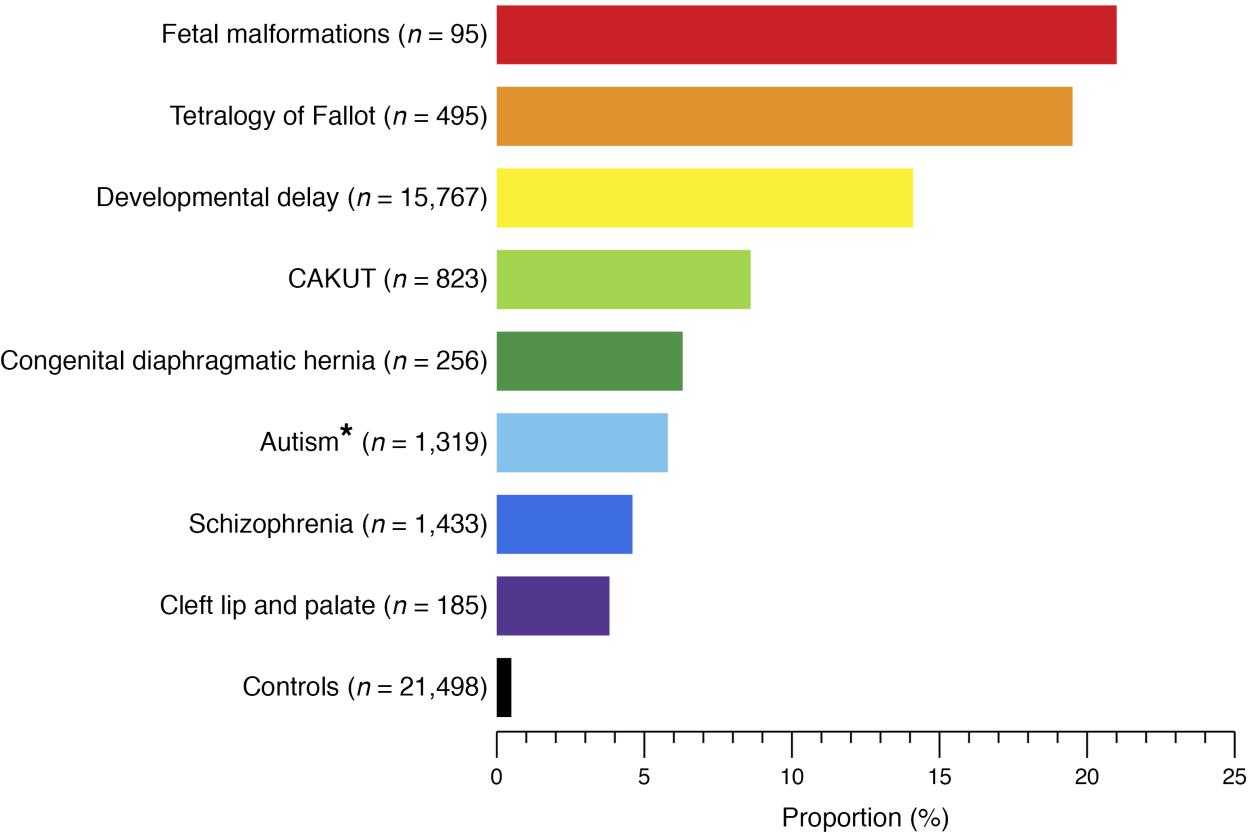
Figure 2 Proportion of patients with known genomic disorders in different human developmental phenotypes and healthy controls.4
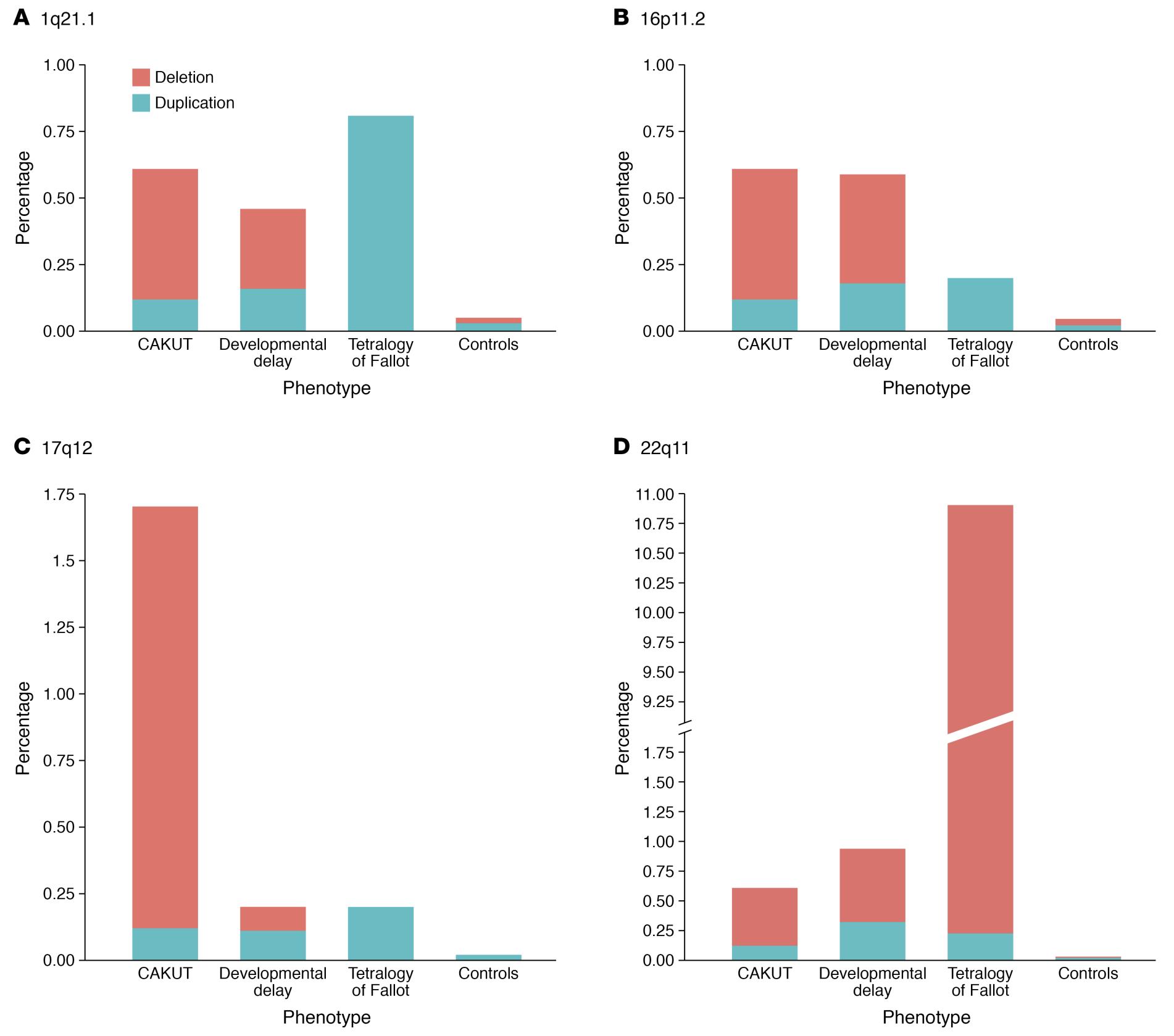
Figure 3 Differences and similarities in the prevalence of the four most commonly implicated CNV loci in CAKUT patients.4
MCDK
MCDK is non-inherited congenital cystic diseases, a type of renal dysplasia that causes multiple non-communicating cysts separated by dysplastic parenchyma. The prevalence of MCDK is about 1 in 3,600 to 1 in 4,300 live births.7,8 The cause of MCDK is unknown, but it is thought to potentially be related to abnormal formation of the ureteric bud, exposure to teratogens, or urinary tract obstruction. MCDK is often detected through antenatal ultrasonography. As it often does not cause symptoms or complications, it is usually only identified postnatally if there is a noticeable mass or if it is discovered incidentally during imaging for another condition.
Some of the genes that have been associated with MCDK include:9,10,11
- HNF1β: This gene encodes a protein called hepatocyte nuclear factor 1 beta, which plays a role in the development and function of the kidney and other organs. Mutations in HNF1β can cause a range of renal anomalies, including MCDK.
- PAX2: This gene encodes a protein called paired box 2, which is important for the development of the ureter, the tube that carries urine from the kidney to the bladder. Mutations in PAX2 can cause MCDK and other renal anomalies.
- SIX1: This gene encodes a protein called sine oculis homeobox 1, which is involved in the development of various organs, including the kidney. Mutations in SIX1 can cause MCDK and other renal anomalies.
- WT1: This gene encodes a protein called Wilms tumor 1, which is important for the development of the kidney and other organs. Mutations in WT1 can cause MCDK and other renal anomalies.
MCDK is typically managed conservatively (i.e., observation) as most cases without any long-term complications. Patients with contralateral abnormalities may experience kidney impairment over time. Thus, long-term follow-up is necessary.
Renal Agenesis
When compared to bilateral renal agenesis, unilateral renal agenesis occurs in one out of every 1000 pregnancies and has a favorable prognosis. It is estimated that between 0.1% and 0.3% of 1,000 births are affected by bilateral renal agenesis, which generally results in poor survival rates. Several inheritance patterns have been described in families with renal agenesis, which may occur as an isolated finding or as part of a syndrome.12
Unilateral renal agenesis can be caused by mutations in several genes, including ATRX, RET, BMP4, FRAS1, FREM1, GRIP1, GLI3, UPK3A, and more. In addition, bilateral renal agenesis can be caused by mutations in the RET, FGF20, ITGA8 genes. Related genes listed on the Table 2.13,14
Renal Dysplasia
Renal hypodysplasia results from a developmental arrest during the first trimester of the fetal life of the metanephric renal blastema. However, it remains unknown what causes renal hypodysplasia; vascular abnormalities in utero as well as genetic development disorders are two possible mechanisms. It is possible for renal hypodysplasia to be an isolated finding or part of a genetic syndrome associated with malformations of the urinary tract. An analysis of developmental genes (HNF1β, PAX2, EYA1, SIX1, and SALL1) involved in renal dysplasia in a large cohort of children with renal hypodysplasia was provided by the ESCAPE Study. According to this study, PAX2 mutations were diagnosed in 15% of patients with renal pathology, and those with genetic deletions or mutations of large genes demonstrated variable renal phenotypes. In addition, 22% of all patients with cystic renal hypodysplasia had a mutation in HNF1, suggesting that individuals with cystic renal dysplasia be screened for HNF1 mutations.15 Renal ultrasonography findings that show reduced renal size and loss of corticomedullary differentiation are clinically indicative of renal hypodysplasia. The development of the end-stage renal disease can develop in some patients with renal hypodysplasia over time. A specific treatment is not available for renal hypodysplasia. It is important to follow up and manage renal functions during years. For patients who develop end-stage renal disease, renal transplantation is the preferred option.
Ureteropelvic Junction
The most common pathological cause of neonatal hydronephrosis is obstruction of the upper part of the ureter. The overall incidence is 1:1,500, with a ratio of 2:1 in newborn males to females.16 An obstruction to the UPJ is usually caused by intrinsic stenosis or, less commonly, by a crossing vessel that causes external compression at the upper part of the ureter. A possible mechanism is that there is an embryological disruption in the proximal ureter that alters the development of circular musculature, by altering collagen fibers and composition between and around the muscular cells.
Several single gene mutations are implicated in UPJO, including Id2, PAX2, EYA, AGTR2, BMP4, SOX17, CHD1L, and DSTYK.17,18 Id2 and Adamts1 are reported to produce a phenotype that is like the human UPJO phenotype. Mice lacking Id2 also exhibit a high ureter insertion into renal pelvis.19
Vesicoureteral Reflux
Several genes have been identified that are associated with the development of VUR. Some of these genes are involved in the development and function of the ureteral smooth muscle. Other genes are involved in the development and function of the bladder. The high prevalence of VUR among twins, siblings, and offspring, suggests a genetic basis for this disease. It has been established that VUR is a genetically heterogeneous disease based on the results of genetic studies in various populations. A genome-wide linkage and association study linked primary non-syndromic VUR to the 10q26 region of the genome.20,21 Other genes that have been associated with the development of VUR include the FOXC1 gene, the MYH11 gene, and the ACTN4 gene. These genes play various roles in the development and function of the urinary tract.22,23 A genome-wide association study using the largest copy number variant analysis and genome-wide association study of VUR is resulting in the identification of five loci which may be associated with VUR, including WDPCP, OTX1, BMP5, WDPCP, and WNT5 with large effects. WNT5 may play a role in the development of the urogenital system. A genome-wide association study using the largest copy number variant analysis and genome-wide association study of VUR is resulting in the identification of five loci which may be associated with VUR, including WDPCP, OTX1, BMP5, WDPCP, and WNT5 with large effects. WNT5 may play a role in the development of the urogenital system.24
Posterior Urethral Valves
A posterior urethral valve is a life-threatening congenital anomaly of the urinary tract that is usually discovered during neonatal development and, despite optimal treatment, results in high rates of renal insufficiency. PUV is estimated to occur in 1 in 7,000–8,000 live births.25
There is a wide range of reports that indicate that PUV is the leading cause of chronic kidney disease in newborn males, and ESRD can occur in a range of 5% to 64% of cases.26,27 A systematic review reported that chronic kidney disease (CKD) and end-stage kidney disease (ESKD) are associated with PUV patients at up to a 32% and 20% risk, respectively.28
To date, few genes have been identified that are associated with posterior urethral valves. Prune Belly syndrome has been linked to HNF1B in only 3% of cases recognized, and most of those diagnosed do not have a genetic cause. Among the CAKUT abnormalities associated with Trisomy 21, posterior urethral valves, pyelectasis, and megaureters are included.29,30
Table 2 A summary of the genes involved in the development of CAKUT.31,32,33,22,23,34,35,36
| Disorder | Genes |
|---|---|
| Renal Hypoplasia/Dysplasia | ACE, AGT, ATRX, CHD7, ESCO2, EYA1, SIX1, FRAS1, FREM2, GATA3, GLI3, GPC3, GRIP1, HNF1β, JAG1, KAL1, LRP4, MKS1–4, NIPBL, PAX2, PBX1, REN, SALL1, SIX5 |
| Renal Agenesis | ATRX, BMP4, CBP/EP300, CHD7, ESCO2, FGF20, FRAS1, FREM2, GATA3, GRIP1, GLI3, HNF1β, ITGA8, JAG1, KAL1, KAL2, RET, SALL1, VANGL1 |
| Cystic dysplasia | CBP/EP300, DHCR7, EVC, EVC2, FRAS1, FREM2, GPC-3, HNF1β, JAG1, KAL1, KAL2, MKS1–4, NS1, PEX, PAX2, WT1 |
| Duplex system | NS1 |
| Horseshoe/Ectopic kidney | GLI3, NIPBL, VANGL1 |
| Hydronephrosis, UPJ | ACE, Adamts-1, AGT, ATRX, BMP4, CHD1L, CHD7, DHCR7, DSTYK, ESCO2, EYA, FRAS1, FREM2, GLI3, HSPG2, JAG1, Id2, NS1, PAX2, PEX, RET, SOX17, VANGL1 |
| Hidroureter, Megaureter | CHD7, GLI3 |
| VUR | ATRX, DHCR7, EYA1, FOX1, GATA3, HOXA13, HPSE2, JAG1, KAL1, KAL2, MYH11, NIPBL, PAX2, SALL1, SIX1, SIX5 |
| PUV | BNC2, SALL1 |
Clinical Importance and Future Implications of Gene Knowledge
As the field of genetic evaluation continues to advance, it is likely that we will see an increasing ability to identify the genetic causes of CAKUT and to predict the likelihood of developing these conditions. This could lead to earlier diagnosis and treatment, which could improve outcomes for affected individuals.
It should be noted that genetic testing is not always necessary for diagnosing CAKUT. A definitive diagnosis is typically made based on a combination of clinical presentation, medical history, and imaging studies. Genetic testing for CAKUT is usually performed in a specialized center under the supervision of a clinical geneticist, as the interpretation of these results can be complex and may have implications for reproductive decisions. Several laboratories (PreventionGenetics, Invitae, Blueprint Genetics, etc.) offer genetic testing for several different CAKUT disorders. Their test panel for CAKUT includes genetic testing for multiple genes that are associated with CAKUT disorders, including HNF1B, NPHP1, and UPK1B. Genetic testing for these disorders can help to confirm a diagnosis, provide information about the severity of the disorder, and aid in genetic counseling and family planning. It is worth noting that other genetic testing companies and laboratories might also have CAKUT as a panel. Since CAKUT is a group of disorders caused by different genetic mutations, the genetic panel test for CAKUT in some companies might not include all the genetic causes of CAKUT, and different laboratories may have a different list of genes covered.
A complete understanding of the genetic components of CAKUT is essential for developing accurate genetic testing strategies. In addition, this understanding will aid in the guidance of clinical decision-making in genetic testing. Including significant clinical outcomes and renal function, longitudinal studies are required to investigate the long-term effects of early diagnosis and management of CAKUT.
Conclusion and Summary
Congenital anomalies of the kidney and urinary tract (CAKUT) are structural abnormalities that affect the kidneys and urinary tract. It is estimated that there are around 4–60 cases per 10,000 births. A CAKUT diagnosis is usually made during a prenatal ultrasound or after birth in newborns with associated clinical signs and symptoms. The most common anomaly is ureteropelvic junction obstruction, which affects approximately 20% of people with the condition. The development of the kidneys and urinary tract involves the formation and differentiation of several structures, including the ureteric buds and metanephros, which must properly connect to form the functional kidneys and urinary tract. Disruptions in this process can lead to CAKUT. There are various categories of CAKUT disorders including those affecting the number, morphology, and position of the kidneys, and those affecting the collecting system. In 10–25% of cases, CAKUT is caused by genetic disorders. Several genes have been identified that are associated with the development of various kidney related diseases. Research is ongoing to improve our understanding of the causes of CAKUT and to develop better prevention and treatment strategies. Despite its current limitations and difficulties, molecular genetic diagnosis may provide hope for improving the clinical management of patients with CAKUT in the future.
Key Points
- Congenital anomalies of the kidney and urinary tract (CAKUT) are a group of structural abnormalities that affect the kidneys and urinary tract, with an estimated 4 to 60 cases per 10,000 births in the overall population.
- The development of the kidneys and urinary tract in the fetus involves a complex process that begins in the early weeks of pregnancy and involves the formation and differentiation of several structures. Disruptions in this process can lead to a range of congenital anomalies.
- The prevalence of CAKUT in preterm infants was 2% in a recent large reported cohort with 409,704 infants.
- The most common anomaly is ureteropelvic junction obstruction, which affects approximately 20% of individuals with the condition.
- In 10-25% of cases, CAKUT is attributed to genetic disorders.
- Genetic evaluation is advancing, increasing the ability to identify the genetic causes of CAKUT and predict the likelihood of developing these conditions.
- Genetic testing is not always necessary for diagnosing CAKUT, it is typically based on a combination of clinical presentation, medical history, and imaging studies.
- A complete understanding of the genetic components of CAKUT is essential for developing accurate genetic testing strategies and guiding clinical decision-making in genetic testing.
- Longitudinal studies are required to investigate the long-term effects of early diagnosis and management of CAKUT.
References
- Murugapoopathy V, Gupta IR. A Primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin J Am Soc Nephrol 2020; 15 (5): 723–731. DOI: 10.2215/cjn.12581019.
- Hays T, Thompson MV, Bateman DA, Sahni R, Tolia VN, Clark RH, et al.. The Prevalence and Clinical Significance of Congenital Anomalies of the Kidney and Urinary Tract in Preterm Infants. JAMA Netw Open 2022; 5 (9): e2231626. DOI: 10.1001/jamanetworkopen.2022.31626.
- Nicolaou N, Renkema KY, Bongers EMHF, Giles RH, Knoers NVAM. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol 2015; 11 (12): 720–731. DOI: 10.1038/nrneph.2015.140.
- Srivastava S, Molinari E, Raman S, Sayer JA. Many Genes–One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front Pediatr 2018; 5. DOI: 10.3389/fped.2017.00287.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. 8 (1): 25–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 2018; 128 (1): 4–15. DOI: 10.1172/jci95300.
- Gordon AC, Thomas DF, Arthur RJ, Irving HC. Multicystic dysplastic kidney: Is nephrectomy still appropriate? J Pediatr Surg 1988; 24 (9): 951. DOI: 10.1016/s0022-3468(89)80673-6.
- Schreuder MF, Westland R, Wijk JAE van. Unilateral multicystic dysplastic kidney: a meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol Dial Transplant 2009; 24 (6): 1810–1818. DOI: 10.1093/ndt/gfn777.
- Kopač M, Kordič R. Associated Anomalies and Complications of Multicystic Dysplastic Kidney. Pediatr Rep 2022; 14 (3): 375–379. DOI: 10.3390/pediatric14030044.
- Fletcher J, Hu M, Berman Y, Collins F, Grigg J, McIver M, et al.. Multicystic Dysplastic Kidney and Variable Phenotype in a Family with a Novel Deletion Mutation ofPAX2. J Am Soc Nephrol 2005; 16 (9): 2754–2761. DOI: 10.1681/asn.2005030239.
- Chang Y-M, Chen C-C, Lee N-C, Sung J-M, Chou Y-Y, Chiou Y-Y. PAX2 Mutation-Related Renal Hypodysplasia: Review of the Literature and Three Case Reports. Front Pediatr 2022; 9 (765929). DOI: 10.3389/fped.2021.765929.
- Bienstock JL, Birsner ML, Coleman F, Hueppchen NA. Successful In Utero Intervention for Bilateral Renal Agenesis. Obstet Gynecol 2014; 124 (2): 413–415. DOI: 10.1097/aog.0000000000000339.
- Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, et al.. Faculty Opinions recommendation of Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2012; 1 (2): 96–200. DOI: 10.3410/f.13934957.15391059.
- Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal Aplasia in Humans Is Associated with RET Mutations. Am J Hum Genet 2008; 82 (2): 344–351. DOI: 10.1016/j.ajhg.2007.10.008.
- Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al.. Faculty Opinions recommendation of Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2006; 7 (10): 864–870. DOI: 10.3410/f.1047767.508721.
- LEBOWITZ ROBERTL, GRISCOM NTHORNE. Neonatal Hydronephrosis. Radiol Clin North Am 1977; 15 (1): 49–59. DOI: 10.1016/s0033-8389(22)02539-8.
- AC SJ, DM M, oes S, AC S. Congenital Anomalies of the Kidney and Urinary Tract: An Overview. Congenital Anomalies of the Kidney and Urinary Tract 2014; 02 (4): 1–13. DOI: 10.1007/978-3-319-29219-9_1.
- Avanoglu A, Tiryaki S. Embryology and Morphological (Mal)Development of UPJ. Front Pediatr 2020; 8: 137. DOI: 10.3389/fped.2020.00137.
- Aoki Y, Mori S, Kitajima K, Yokoyama O, Kanamaru H, Okada K, et al.. Id2 haploinsufficiency in mice leads to congenital hydronephrosis resembling that in humans. Genes Cells 2004; 9 (12): 1287–1296. DOI: 10.1111/j.1365-2443.2004.00805.x.
- Williams G, Fletcher JT, Alexander SI, Craig JC. Vesicoureteral reflux. Journal of the American Society of Nephrology 2008; 19 (5): 847–862. DOI: 10.1681/asn.2007020245.
- Darlow JM, Darlay R, Dobson MG, Stewart A, Charoen P, Southgate J, et al.. Genome-wide linkage and association study implicates the 10q26 region as a major genetic contributor to primary nonsyndromic vesicoureteric reflux. Sci Rep 2017; 7 (1): 59. DOI: 10.1038/s41598-017-15062-9.
- Woolf AS, Lopes FM, Ranjzad P, Roberts NA. Congenital Disorders of the Human Urinary Tract: Recent Insights From Genetic and Molecular Studies. Front Pediatr 2019; 7 (136). DOI: 10.3389/fped.2019.00136.
- Wu C-HW, Mann N, Nakayama M, Connaughton DM, Dai R, Kolvenbach CM, et al.. Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT). Genet Med 2020; 22 (10): 1673–1681. DOI: 10.1038/s41436-020-0844-z.
- Verbitsky M, Krithivasan P, Batourina E, Khan A, Graham SE, Marasà M, et al.. Review of: "Genome-wide association study followed by trans-ancestry meta-analysis identify 17 new risk loci for schizophrenia". J Am Soc Nephrol 2021; 32 (4): 805–820. DOI: 10.32388/jqb9pr.
- Thakkar D, Deshpande AV, Kennedy SE. Epidemiology and demography of recently diagnosed cases of posterior urethral valves. Pediatr Res 2014; 76 (6): 560–563. DOI: 10.1038/pr.2014.134.
- Warshaw BL, Edelbrock HH, Ettenger RB, Malekzadeh MH, Pennisi AJ, Uittenbogaart CH, et al.. Renal transplantation in children with obstructive uropathy. J Pediatr Surg 1980; 15 (6): 986. DOI: 10.1016/s0022-3468(80)80355-1.
- Roth KS, Carter WH, Chan JCM. Obstructive Nephropathy in Children: Long-Term Progression After Relief of Posterior Urethral Valve. Pediatrics 2001; 107 (5): 1004–1010. DOI: 10.1542/peds.107.5.1004.
- Hennus PML, Kort LMO de, Bosch JLH, Jong TPVM de, Heijden GJMG van der. A Systematic Review on the Accuracy of Diagnostic Procedures for Infravesical Obstruction in Boys. PLoS One 2014; 9 (2): e85474. DOI: 10.1371/journal.pone.0085474.
- Rodriguez MM. Congenital anomalies of the kidney and urinary tract (CAKUT). Lijec Vjesn 2014; 144 (Supp 1). DOI: 10.26800/lv-144-supl1-26.
- Postolache L, Parsa A, Simoni P, Boitsios G, Ismaili K, Schurmans T, et al.. Widespread kidney anomalies in children with Down syndrome. Pediatr Nephrol 2022; 37 (10): 2361–2368. DOI: 10.1007/s00467-022-05455-y.
- Uy N, Reidy K. Developmental Genetics and Congenital Anomalies of the Kidney and Urinary Tract. J Pediatr Genet 2016; 05 (01): 051–060. DOI: 10.1055/s-0035-1558423.
- Kolvenbach CM, Dworschak GC, Frese S, Japp AS, Schuster P, Wenzlitschke N, et al.. Expanding congenital abnormalities of the kidney and urinary tract (CAKUT) genetics: basonuclin 2 (BNC2) and lower urinary tract obstruction. Ann Transl Med 2019; 7 (S6): S226–s226. DOI: 10.21037/atm.2019.08.73.
- Heidet L, Morinière V, Henry C, De Tomasi L, Reilly ML, Humbert C, et al.. Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 2017; 28 (10): 2901–2914. DOI: 10.1681/asn.2017010043.
- Talati AN, Webster CM, Vora NL. Prenatal genetic considerations of congenital anomalies of the kidney and urinary tract (CAKUT). Prenat Diagn 2019; 39 (9): 679–692. DOI: 10.1002/pd.5536.
- Puri P, Gosemann J-H, Darlow J, Barton DE. Genetics of vesicoureteral reflux. Nat Rev Urol 2011; 8 (10): 539–552. DOI: 10.1038/nrurol.2011.113.
- Chen F. Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol 2009; 24 (9): 1621–1632. DOI: 10.1007/s00467-008-1072-y.
Last updated: 2025-09-25 12:10