
37: Épispadias féminin
Ce chapitre prendra environ 16 minutes de lecture.
Introduction
L’exstrophie vésicale (EV) est une anomalie congénitale dévastatrice de l’appareil urinaire dans laquelle les nourrissons naissent avec la vessie extériorisée à travers la paroi abdominale, un urètre ouvert dorsalement, et d’importantes anomalies secondaires des organes génitaux. Elle est largement considérée comme l’affection congénitale de l’appareil urinaire la plus difficile et la plus complexe sur le plan chirurgical.1 L’EV varie en degré de sévérité et s’inscrit sur un continuum décrit comme le complexe exstrophie-épispadias (CEE).2 L’incidence du CEE est ~1 sur 10 000.2
L’exstrophie cloaquale (CE) se situe à l’extrémité sévère du spectre du BEEC. La CE est une maladie congénitale rare, avec une incidence de 1 sur 200 000–400 000 naissances vivantes.3,4 Les anomalies associées fréquentes comprennent l’omphalocèle, l’exstrophie vésicale, l’anus imperforé et le dysraphisme spinal.4 Les caractéristiques supplémentaires comprennent une plaque médiane de cæcum exstrophié encadrée par 2 hémivésiques, un segment d’iléon prolabant à partir de la plaque cæcale et un intestin postérieur rudimentaire se terminant en cul-de-sac, associé à un anus imperforé.4
À l’autre extrémité du spectre se trouve l’épispadias. L’épispadias est le plus souvent présent comme composante de la BE et de la CE, mais lorsque l’épispadias survient isolément, en l’absence de BE et de CE, il est considéré comme la variante la moins sévère du BEEC. L’épispadias est défini par une fusion incomplète de l’urètre dorsal.5 Cela va d’un défaut glandulaire léger dans un pénis couvert jusqu’à la forme pénopubienne qui s’étend proximalement jusqu’au col vésical et entraîne une incontinence complète chez les hommes comme chez les femmes.5 Fait intéressant, le premier cas décrit d’épispadias est attribué à l’empereur byzantin Héraclius (CE 610–641).6 L’épispadias masculin isolé est une anomalie rare, avec une incidence rapportée de 1 sur 117 000 hommes.6 L’épispadias féminin isolé est encore plus rare, avec une incidence de 1 sur 484 000 patientes.6 Dans ce chapitre, nous nous concentrerons exclusivement sur l’épispadias féminin.
En 1928, Davis a classé l’épispadias féminin en trois catégories selon la sévérité. Ce système de classification est encore utilisé aujourd’hui.7 Dans les formes mineures d’épispadias, l’orifice urétral apparaît simplement béant. Dans l’épispadias intermédiaire, l’urètre est fendu dorsalement sur la majeure partie de sa longueur. Dans la forme la plus sévère d’épispadias, la fente urétrale intéresse toute la longueur de l’urètre et le mécanisme sphinctérien est rendu incompétent. À noter que la véritable prévalence de l’épispadias féminin pourrait être plus élevée que ce qui est rapporté, car dans les formes moins sévères, les modifications des organes génitaux externes peuvent être minimes et, en l’absence d’incontinence, les patientes ne sont pas diagnostiquées.8,9
Embryologie
L’une des principales théories du maldéveloppement embryonnaire dans l’exstrophie, proposée par Marshall et Muecke, est que le défaut de base est un surdéveloppement anormal de la membrane cloacale au cours de la quatrième semaine de gestation, ce qui empêche la migration médiale du tissu mésenchymateux et le développement correct de la paroi abdominale inférieure. On pense que le moment de la rupture de cette membrane cloacale défectueuse détermine la variante résultante.10 On pense qu’il existe un défaut de renforcement de la membrane cloacale par l’envahissement du mésoderme. La membrane cloacale est une couche bilaminaire située à l’extrémité caudale du disque germinatif qui occupe la paroi abdominale infra-ombilicale. L’envahissement mésenchymateux entre les couches ectodermique et endodermique de la membrane cloacale aboutit à la formation des muscles de la paroi abdominale inférieure et des os pelviens. La membrane cloacale est sujette à une rupture prématurée, et si cela se traduit par un petit défaut infra-ombilical, ou si la rupture survient à un stade plus tardif du développement, on pense qu’il en résulte un épispadias, tandis que dans les cas de défauts plus étendus ou de rupture plus précoce, BE ou CE peuvent en résulter.10,11
Il existe d’autres théories plausibles concernant la cause du complexe exstrophie-épispadias. Développement anormal des éminences génitales caudalement par rapport à la position normale, avec fusion sur la ligne médiane
en dessous plutôt qu’au-dessus de la membrane cloaquale, a été adoptée par d’autres chercheurs.11,12 Une autre hypothèse intéressante, encore controversée, décrit une insertion caudale anormale du pédicule embryonnaire, qui entraîne un défaut d’interposition du tissu mésenchymateux au niveau de la ligne médiane.13 En conséquence de ce défaut, la translocation du cloaque vers les profondeurs de la cavité abdominale ne se produit pas. Une membrane cloaquale qui demeure en position infra-ombilicale superficielle représente un état embryonnaire instable avec une forte tendance à se désintégrer, ce qui a été étayé par les travaux de laboratoire de Thomalla et al.14,15 Une autre théorie intéressante est qu’il s’agit plutôt d’un développement anormal du bassin osseux qui constitue le facteur déclenchant du développement de l’exstrophie. Beaudoin et al ont suggéré que l’absence de “rotation” du primordium de l’anneau pelvien empêche les structures attachées à l’anneau pelvien de se rejoindre au niveau de la ligne médiane, permettant ainsi la survenue d’une hernie vésicale.16
Épidémiologie
L’épispadias féminin complet est une malformation congénitale extrêmement rare, survenant dans environ 1/484 000 naissances vivantes, beaucoup moins fréquemment que l’EV, dont l’incidence est estimée entre 1/10 000 et 1/50 000.6 Le rapport hommes-femmes de l’exstrophie vésicale, d’après plusieurs séries, est de 2,3:1, et l’épispadias féminin est également moins fréquent que l’épispadias masculin, dont l’incidence rapportée est de 1 pour 117 000 sujets de sexe masculin.17
Le risque de récurrence de la BE dans une famille donnée est d’environ 1 sur 100.18 Shapiro et al ont, dans un questionnaire, identifié la récurrence de la BE et de l’épispadias dans seulement 9 des environ 2 500 cas indexés.19 Shapiro et al ont déterminé que le risque de BE chez la descendance d’individus atteints de BE et d’épispadias est de 1 sur 70 naissances vivantes, soit une incidence 500 fois plus élevée que dans la population générale.17
Pathogénie
Notre compréhension actuelle de la pathogenèse de l’épispadias est incomplète. En raison du manque de recherches de qualité, les experts ne sont toujours pas sûrs des origines embryologiques et des mécanismes génétiques à l’origine de l’épispadias. Cependant, des progrès ont été réalisés ces dernières années en ce qui concerne l’identification de gènes candidats dans la BEEC, ce qui engloberait également le développement de l’épispadias isolé. von Lowtow et al ont évalué 169 personnes atteintes de BEEC (recrutées majoritairement en Europe, origine génétique géo-ethnique non rapportée).20 Ils ont identifié plusieurs individus présentant des variations du nombre de copies pathogènes, dont un avec une duplication précédemment rapportée en 22q11. Cette région est également associée au syndrome de délétion 22q11 (syndrome de DiGeorge), et CAKUT.21 Une méta-analyse ultérieure d’études d’association pangénomique (GWAS) combinant 568 patients BEEC et 3,241 témoins d’origine européenne a identifié une association avec un locus contenant l’enhancer transcriptionnel ISL1 (p = 2.22 × 10−08).22,23,24,25,26 D’autres études fonctionnelles et sur modèles ont renforcé un rôle causal possible d’ISL1 dans la BEEC. Par exemple, des modèles de biologie du développement ont été utilisés pour préciser la localisation de l’activité d’ISL1 dans l’appareil urinaire en formation.26 L’analyse de lignée génétique des cellules exprimant ISL1 au moyen d’un modèle murin traceur de lignée a montré des cellules exprimant ISL1 dans les voies urinaires d’embryons de souris.26
Évaluation et diagnostic
Même avec les modalités modernes d’échographie, le diagnostic prénatal de la BE est souvent difficile à préciser, et le diagnostic prénatal de l’épispadias isolé est presque impossible.27 En 2012, Goyal et al. ont constaté que seulement 25% des cas de BEEC sont diagnostiqués en prénatal.27 Plusieurs groupes ont tenté de définir des critères importants pour le diagnostic prénatal de la BE, mais il n’existe pas de caractéristiques spécifiques pour l’épispadias. Ils ont constaté que, pour la BE classique, l’absence d’une vessie normalement remplie de liquide lors d’examens répétés suggère le diagnostic, tout comme la présence d’une masse de tissu échogène sur la paroi abdominale inférieure.28,29 Une longueur entre l’insertion du cordon ombilical et le tubercule génital inférieure au 5e percentile pour l’âge gestationnel fournit également une mesure quantitative permettant d’évaluer un diagnostic de BE.30 Dans une revue de 25 échographies prénatales avec la naissance ultérieure d’un nouveau-né atteint de BE, Gearhart et al ont fait plusieurs observations:1 absence de remplissage vésical,2 un ombilic bas situé,3 un élargissement des branches pubiennes,4 des organes génitaux de petite taille, et5 une masse de la paroi abdominale inférieure qui augmentait de taille au fur et à mesure de la progression de la grossesse et de l’augmentation de volume des viscères intra-abdominaux.31 Il n’est pas difficile de comprendre pourquoi le diagnostic prénatal de l’épispadias isolé est plus difficile. Parmi les observations prénatales mentionnées précédemment, seuls un ombilic bas situé, un élargissement des branches pubiennes et des organes génitaux de petite taille pourraient potentiellement être présents dans le cadre d’un épispadias isolé. Tous ces signes seraient moins marqués dans le cadre d’un épispadias isolé, et la notion d’organes génitaux de petite taille ne s’appliquerait pas dans le cas d’un épispadias isolé chez la fille.
Par conséquent, le diagnostic d’épispadias féminin isolé est généralement posé par examen physique après la naissance. Les modifications de l’apparence externe des organes génitaux peuvent être minimes, en particulier dans les cas d’épispadias léger. Sur le plan externe, on mettra en évidence une malformation génitale caractérisée par un clitoris bifide. Le mont du pubis aura une forme déprimée et sera recouvert d’une zone cutanée lisse et glabre. Sous cette zone, il peut exister une quantité modérée de tissu sous-cutané et de graisse, ou bien la peau peut se trouver directement en avant et en dessous de la surface de la symphyse pubienne. Les petites lèvres sont habituellement peu développées et se terminent en avant au niveau de l’hémi-clitoris correspondant du clitoris bifide, où il peut exister un rudiment de repli préputial. La symphyse pubienne est habituellement soudée, mais peut être séparée par une étroite bande fibreuse. À l’écartement des lèvres, l’anomalie urétrale est identifiée et peut varier considérablement selon le degré d’épispadias présent. L’anomalie urétrale peut aller d’un défaut distal court à une atteinte s’étendant proximalement jusqu’au col vésical (Figure 1). Le vagin et les organes génitaux internes sont habituellement normaux. Du fait que les modifications de l’aspect externe peuvent être minimes, certains enfants ne sont identifiés qu’en raison d’une incontinence persistante et, dans les formes légères sans incontinence, le diagnostic peut ne jamais être posé. C’est pourquoi la prévalence réelle de l’épispadias féminin pourrait être plus élevée que ce qui est rapporté.8,9 Ainsi, l’épispadias féminin isolé comporte toujours une anomalie de l’urètre, mais il peut coexister des anomalies du col vésical qu’il ne faut pas négliger lors de la prise en charge de ces patientes. Dans les formes plus sévères d’épispadias féminin, la vessie peut être petite et comparable à celle de patients présentant une exstrophie vésicale refermée, puisqu’il n’y a pas eu de cycles vésicaux in utero. En fait, un tiers de tous les patients porteurs d’épispadias avec incontinence ont une capacité vésicale inférieure à 60 mL et l’incidence du reflux vésico-urétéral est de 30 % à 75 %.32
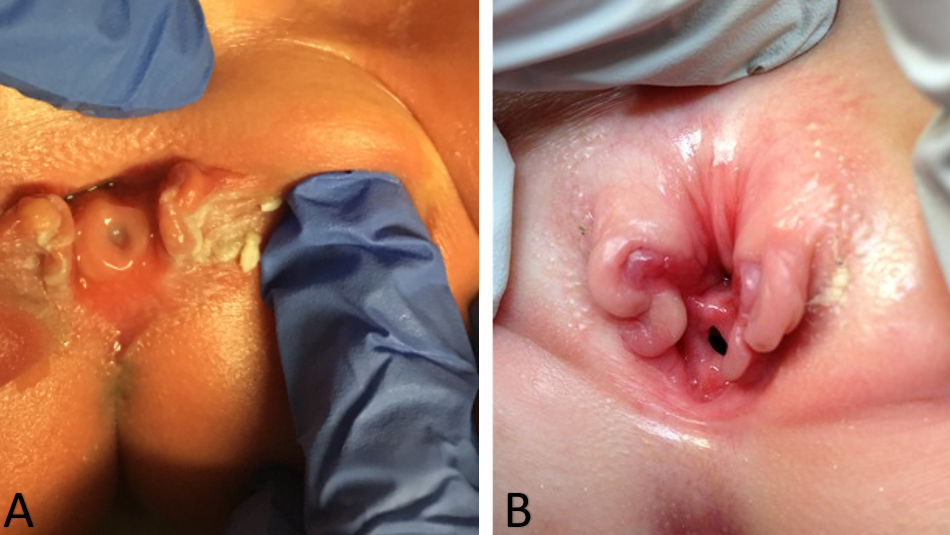
Figure 1 Cas sévères d’épispadias féminin. Épispadias pénopubien chez un nouveau-né (A). Épispadias pénopubien chez un enfant de 4 ans (B).
Traitement et résultats
Chez la patiente présentant un épispadias féminin, les objectifs du traitement sont l’obtention de la continence urinaire, la préservation des voies urinaires supérieures et la reconstruction d’organes génitaux externes fonctionnels et acceptables sur le plan esthétique. Chez les patientes présentant un épispadias léger qui sont continentes et n’ont que des modifications minimes de leurs organes génitaux externes, une reconstruction chirurgicale peut ne pas être nécessaire. Pour les formes plus sévères d’épispadias féminin, une reconstruction chirurgicale est nécessaire pour obtenir la continence et corriger les anomalies des organes génitaux externes.
Historiquement, de nombreuses opérations ont été rapportées pour tenter d’obtenir la continence chez les patients atteints d’épispadias, mais les résultats étaient décevants. Ces procédures comprenaient la plicature transvaginale de l’urètre et du col vésical, des transplantations musculaires, la torsion de l’urètre, la cautérisation de l’urètre, un lambeau vésical et la suspension vésico-urétrale de Marshall-Marchetti.33,34,35,36 Ces procédures visaient à augmenter la résistance urétrale, mais n’ont pas corrigé l’incontinence, car elles n’ont pas cherché à normaliser l’anatomie de l’urètre et du col vésical.
Comme mentionné précédemment, les patients présentant un épispadias distal sans incontinence peuvent ne nécessiter aucune intervention chirurgicale. En cas d’épispadias féminin proximal, nous pratiquons souvent la réparation de l’épispadias et la reconstruction du col vésical, avec ou sans ostéotomies, dans le même temps opératoire (Figure 2). Cela est similaire à la réparation primaire complète de l’exstrophie que nous réalisons dans les cas de BE et est généralement effectué entre 3–6 mois de vie. Nous avons constaté que la résistance supplémentaire procurée par la réparation de l’épispadias et la reconstruction du col vésical permet un cyclage vésical et peut souvent se traduire par une augmentation de la capacité vésicale. Ces constatations ont été corroborées par d’autres auteurs qui ont montré que la fermeture primaire de l’urètre épispadique chez des enfants atteints d’exstrophie fermée augmentait la capacité vésicale sans provoquer d’hydronéphrose. Cette approche a été appliquée à des patients de sexe masculin et féminin présentant un épispadias.37,38,39
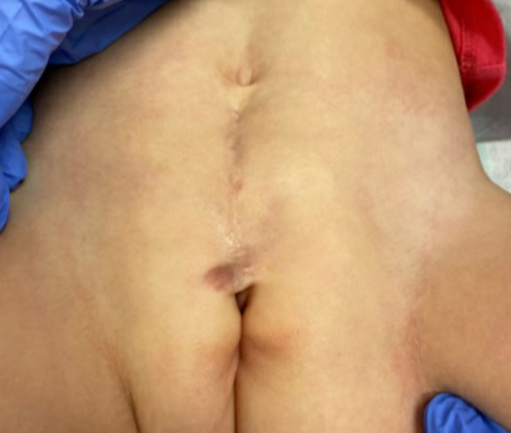
Figure 2 Image postopératoire d’une patiente présentant un épispadias féminin sévère, après réparation de l’épispadias et reconstruction du col vésical.
Les taux de continence chez les patientes présentant un épispadias isolé ont été estimés entre 67 et 87,5 %.6,32,40 L’intervalle moyen entre la reconstruction et l’obtention de la continence a été rapporté entre 18 et 30 mois.6,41 Il existe également de rares rapports de patientes présentant une acquisition tardive d’une continence complète plusieurs années après la reconstruction.32 Ce retard prolongé dans l’obtention de la continence serait secondaire à un développement musculaire pelvien accru au fil du temps, et en particulier après les changements hormonaux de la puberté.32
Complications
La prise en charge chirurgicale de l’épispadias est notoirement difficile et, par conséquent, les complications ne sont pas rares. Les complications potentielles après la réparation de l’urètre épispadique comprennent la fistule urétrocutanée, la sténose méatique et les sténoses urétrales. Chez les patients nécessitant une intervention sur le col vésical, l’importance d’un suivi étroit ne saurait être trop soulignée. Lorsque la résistance de sortie de la vessie est augmentée, les patients courent le risque de développer une vessie à haute pression, la vessie se contractant pour lutter contre cette résistance élevée, ce qui expose leurs voies urinaires supérieures à un risque. Il est donc crucial que les chirurgiens assurent un suivi très rapproché après la reconstruction afin d’identifier précocement l’apparition d’une hydronéphrose ou d’autres signes d’un système vésical à haute pression. Cela est particulièrement important chez les patients qui présentaient, en préopératoire, des vessies de petite capacité et peu complianteS, bien que cela soit très rare dans l’épispadias. En outre, après une intervention sur le col vésical, les patients peuvent encore souffrir d’incontinence et nécessiter des interventions supplémentaires pour obtenir la continence.
Chez les patients présentant des formes sévères d’épispadias proximal, des ostéotomies iliaques bilatérales peuvent être nécessaires au moment de la reconstruction urologique afin de diminuer la tension sur la fermeture en réduisant le diastasis pubien. Les risques associés aux ostéotomies iliaques bilatérales comprennent la perte sanguine, la diminution de la perfusion des membres inférieurs et, dans les cas très sévères, la diminution de la perfusion du clitoris. La diminution de la perfusion du clitoris peut survenir lorsque les os du pubis sont rapprochés trop étroitement après les ostéotomies. Des fistules vésico-cutanées sont également possibles chez les patientes qui nécessitent une reconstruction du col vésical. Fait intéressant, lorsque Suson et al ont comparé 22 filles présentant un épispadias complet à 23 filles avec une BE classique, ils n’ont pas trouvé de différence quant au nombre d’interventions chirurgicales, à l’acquisition de la continence avec ou sans nécessité de reconstruction du col vésical, ou au besoin ultérieur d’une dérivation urinaire continente.5
Suivi recommandé
Les patients présentant un épispadias distal qui urinent normalement et dont les voies urinaires supérieures sont saines peuvent souvent faire l’objet d’une simple surveillance et, éventuellement, être sortis de la prise en charge après une période de suivi. À l’inverse, les patients présentant des formes plus sévères d’épispadias ayant subi une reconstruction doivent être suivis tout au long de l’enfance et jusqu’à l’âge adulte. Il est essentiel de transférer, à l’âge adulte, leur prise en charge à un urologue spécialisé en médecine de transition, ayant l’expérience du traitement de patients présentant des anomalies urologiques congénitales complexes.
Comme mentionné précédemment, les patients qui subissent une réparation de l’épispadias et une reconstruction du col vésical nécessitent un suivi postopératoire très étroit. Des études ont montré que les patients qui subissent des interventions isolées sur le col vésical sans agrandissement vésical concomitant sont exposés au risque de développer des pressions vésicales élevées et, par la suite, une détérioration des voies urinaires supérieures. Cela a été observé même lorsque des bilans préopératoires approfondis sont réalisés afin de garantir des pressions vésicales et une compliance vésicale préopératoires jugées sûres. Malgré la sélection préopératoire, des modifications significatives de la dynamique vésicale surviennent, et 40 % des patients ayant une vessie neurologique ont nécessité un agrandissement vésical secondaire.42 Ainsi, un suivi annuel avec des échographies réno-vésicales est absolument indispensable chez ces patients.
Conclusions
L’épispadias féminin isolé est une affection extrêmement rare et est considéré comme la variante la moins sévère du BEEC. Elle varie d’un défaut distal léger qui n’affecte pas la continence, à un phénotype complet qui s’étend proximalement jusqu’au col vésical et entraîne une incontinence complète.5 Comme pour la BE, l’épispadias proximal est largement considéré comme l’un des troubles congénitaux des voies urinaires les plus difficiles sur le plan chirurgical et les plus complexes.1 Une réparation de l’épispadias, une reconstruction du col vésical, et même des ostéotomies peuvent être nécessaires dans les formes les plus sévères. De nombreux patients atteints d’épispadias nécessiteront un suivi à vie par un urologue ayant l’expérience de la prise en charge de patients présentant des anomalies urologiques complexes.
Points clés
- L’épispadias est la forme la moins sévère du BEEC et se définit par une fusion incomplète de l’urètre dorsal.
- L’épispadias féminin complet est une malformation congénitale extrêmement rare, à raison d’environ 1/484,000 naissances vivantes, beaucoup moins fréquente que la BE, dont l’incidence est estimée à 1/10,000 à 1/50,000.
- Le diagnostic d’épispadias féminin isolé est généralement posé par l’examen clinique en postnatal, car le diagnostic par imagerie prénatale n’est pas fiable.
- L’anomalie urétrale peut aller d’un court défaut distal à un défaut qui s’étend proximalement jusqu’au col vésical
- Chez les patientes atteintes d’épispadias féminin, les objectifs du traitement sont l’obtention de la continence urinaire, la préservation des voies urinaires supérieures et la reconstruction d’organes génitaux externes fonctionnels et esthétiquement acceptables.
- Chez les patientes présentant un épispadias léger qui sont continentes et n’ont que des modifications minimes de leurs organes génitaux externes, une reconstruction chirurgicale peut ne pas être nécessaire.
- Pour les formes plus sévères d’épispadias féminin, une reconstruction chirurgicale est nécessaire pour obtenir la continence et corriger les anomalies des organes génitaux externes.
- Les taux de continence chez les femmes présentant un épispadias isolé ont été rapportés entre 67-87.5%.
Références
- Weiss DA, Shukla AR, Borer JG, Sack BS, Kryger JV, Roth EB, et al.. Evaluation of outcomes following complete primary repair of bladder exstrophy at three individual sites prior to the establishment of a multi-institutional collaborative model. J Pediatr Urol 2020; 16 (4): 435.e1–435.e6. DOI: 10.1016/j.jpurol.2020.05.153.
- Reutter H, Keppler-Noreuil K, E. Keegan C, Thiele H, Yamada G, Ludwig M. Genetics of Bladder-Exstrophy-Epispadias Complex (BEEC): Systematic Elucidation of Mendelian and Multifactorial Phenotypes. Curr Genomics 2005; 17 (1): 4–13. DOI: 10.2174/1389202916666151014221806.
- Phillips TM. Spectrum of cloacal exstrophy. Semin Pediatr Surg 2011; 20 (2): 113–118. DOI: 10.1053/j.sempedsurg.2010.12.007.
- Woo LL, Thomas JC, Brock JW. Cloacal exstrophy: A comprehensive review of an uncommon problem. J Pediatr Urol 2010; 6 (2): 102–111. DOI: 10.1016/j.jpurol.2009.09.011.
- Suson KD, Preece J, Baradaran N, Di Carlo HN, Gearhart JP. The Fate of the Complete Female Epispadias and Exstrophy Bladder–Is There a Difference? J Urol 2013; 190 (4s): 1583–1589. DOI: 10.1016/j.juro.2013.01.093.
- Gearhart JP. Exstrophy-Epispadias Complex in Campbell-Walsh-Wein Urology 2021. Elsevier; .
- Davis DM. Epispadias in Females and Its Surgical Treatment. J Urol 1928; 20 (6): 673–678. DOI: 10.1016/s0022-5347(17)73196-5.
- Yeni E, Unal D, Verit A, Karatas OF. An adult female epispadias without exstrophy was presented with urinary incontinence: a case report. Int Urogynecol J 2004; 15 (3): 212–213. DOI: 10.1007/s00192-004-1131-2.
- Krishna Shetty MV, Sen TK, Bhaskaran VA. Female epispadias. Afr J Paediatr Surg 2011; 8 (2): 215. DOI: 10.4103/0189-6725.86066.
- Muecke EC. The Role of the Cloacae Membrane in Exstrophy: The First Successful Experimental Study. J Urol 1964; 92 (6): 659–668. DOI: 10.1016/s0022-5347(17)64028-x.
- Ambrose SS, O’Brien DP. Surgical Embryology of the Exstrophy-Epispadias Complex. Surg Clin North Am 1974; 54 (6): 1379–1390. DOI: 10.1016/s0039-6109(16)40493-7.
- Patten BM, Barry A. The genesis of exstrophy of the bladder and epispadias. Am J Anat 1952; 90 (1): 35–57. DOI: 10.1002/aja.1000900103.
- Mildenberger H, Kluth D, Dziuba M. Embryology of bladder exstrophy. J Pediatr Surg 1988; 23 (2): 166–170. DOI: 10.1016/s0022-3468(88)80150-7.
- Johnston JH, Kogan SJ. The exstrophic anomalies and their surgical reconstruction. Curr Probl Surg 1974; 11 (8): 1–39. DOI: 10.1016/s0011-3840(74)80011-0.
- Thomalla JV, Rudolph RA, Rink RC, Mitchell ME. Induction of Cloacal Exstrophy in the Chick Embryo Using the CO 2 Laser. J Urol 1985; 134 (5): 991–995. DOI: 10.1016/s0022-5347(17)47573-2.
- Beaudoin S, Simon L, Bargy F. Anatomical basis of a common embryological origin for epispadias and bladder or cloacal exstrophies. Surg Radiol Anat 1997; 19 (1): 11–16. DOI: 10.1007/bf01627728.
- Shapiro E, Lepor H, Jeffs RD. The Inheritance of the Exstrophy-Epispadias Complex. J Urol 1984; 132 (2): 308–310. DOI: 10.1016/s0022-5347(17)49605-4.
- Ives E, Coffey R, Carter CO. A family study of bladder exstrophy. J Med Genet 1980; 17 (2): 139–141. DOI: 10.1136/jmg.17.2.139.
- Shapiro E, Jeffs RD, Gearhart JP, Lepor H. Muscarinic Cholinergic Receptors in Bladder Exstrophy: Insights Into Surgical Management. J Urol 1985; 134 (2): 308–310. DOI: 10.1016/s0022-5347(17)47139-4.
- Reutter H, Boyadjiev SA, Gambhir L, Ebert A-K, Rösch WH, Stein R, et al.. Phenotype Severity in the Bladder Exstrophy-Epispadias Complex: Analysis of Genetic and Nongenetic Contributing Factors in 441 Families from North America and Europe. J Pediatr 2011; 159 (5): 825–831.e1. DOI: 10.1016/j.jpeds.2011.04.042.
- WOOD HADLEYM, TROCK BRUCEJ, GEARHART JOHNP. In Vitro Fertilization and the Cloacal-Bladder Exstrophy-Epispadias Complex: Is there an Association? J Urol 2003; 169 (4): 1512–1515. DOI: 10.1097/01.ju.0000054984.76384.66.
- Lowtzow C von, Hofmann A, Zhang R, Marsch F, Ebert A-K, Rösch W, et al.. CNV analysis in 169 patients with bladder exstrophy-epispadias complex. BMC Med Genet 2016; 17 (1): 35. DOI: 10.1186/s12881-016-0299-x.
- Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, et al.. Copy-Number Disorders Are a Common Cause of Congenital Kidney Malformations. Am J Hum Genet 2012; 91 (6): 987–997. DOI: 10.1016/j.ajhg.2012.10.007.
- Arkani S, Cao J, Lundin J, Nilsson D, Källman T, Barker G, et al.. Evaluation of the ISL1 gene in the pathogenesis of bladder exstrophy in a Swedish cohort. Hum Genome Var 2018; 5 (1): 18009. DOI: 10.1038/hgv.2018.9.
- Draaken M, Knapp M, Pennimpede T, Schmidt JM, Ebert A-K, Rösch W, et al.. Genome-wide Association Study and Meta-Analysis Identify ISL1 as Genome-wide Significant Susceptibility Gene for Bladder Exstrophy. PLoS Genet 2015; 11 (3): e1005024. DOI: 10.1371/journal.pgen.1005024.
- Reutter H, Draaken M, Pennimpede T, Wittler L, Brockschmidt FF, Ebert A-K, et al.. Genome-wide association study and mouse expression data identify a highly conserved 32 kb intergenic region between WNT3 and WNT9b as possible susceptibility locus for isolated classic exstrophy of the bladder. Hum Mol Genet 2014; 23 (20): 5536–5544. DOI: 10.1093/hmg/ddu259.
- Reutter H, Hoischen A, Ludwig M, Stein R, Radlwimmer B, Engels H, et al.. Genome-wide analysis for micro-aberrations in familial exstrophy of the bladder using array-based comparative genomic hybridization. BJU Int 2007; 100 (3): 646–650. DOI: 10.1111/j.1464-410x.2007.07086.x.
- Zhang R, Knapp M, Suzuki K, Kajioka D, Schmidt JM, Winkler J, et al.. ISL1 is a major susceptibility gene for classic bladder exstrophy and a regulator of urinary tract development. Sci Rep 2017; 7 (1): 42170. DOI: 10.1038/srep42170.
- Goyal A, Fishwick J, Hurrell R, Cervellione RM, Dickson AP. Antenatal diagnosis of bladder/cloacal exstrophy: Challenges and possible solutions. J Pediatr Urol 2012; 8 (2): 140–144. DOI: 10.1016/j.jpurol.2011.05.003.
- Mirk P, Calisti A, Fileni A. Prenatal sonographic diagnosis of bladder extrophy. J Ultrasound Med 1986; 5 (5): 291–293. DOI: 10.7863/jum.1986.5.5.291.
- VERCO PW, KHOR BH, BARBARY J, ENTHOVEN C. Ectopia Vesicae in Utero. Australas Radiol 1986; 30 (2): 117–120. DOI: 10.1111/j.1440-1673.1986.tb02400.x.
- Fishel-Bartal M, Perlman S, Messing B, Bardin R, Kivilevitch Z, Achiron R, et al.. Early Diagnosis of Bladder Exstrophy: Quantitative Assessment of a Low-Inserted Umbilical Cord. J Ultrasound Med 2017; 36 (9): 1801–1805. DOI: 10.1002/jum.14212.
- GEARHART J, BENCHAIM J, JEFFS R, SANDERS R. Criteria for the prenatal diagnosis of classic bladder exstrophy. Obstet Gynecol 1995; 85 (6): 961–964. DOI: 10.1016/0029-7844(95)00069-4.
- Kelalis PP, Kramer SA. Surgical Correction of Female Epispadias. Eur Urol 1982; 8 (6): 321–324. DOI: 10.1159/000473547.
- Jonuzi A, Popović N, Zvizdić Z, Milišić E, Karavdić K, Dewan P. Female Epispadias Presenting as Urinary Incontinence. APSP J Case Rep 2017; 8 (2): 10. DOI: 10.21699/ajcr.v8i2.548.
- Gross RE, Cresson SL. Exstrophy Of Bladder. J Am Med Assoc 1952; 149 (18): 1640. DOI: 10.1001/jama.1952.02930350028008.
- MARSHALL VICTORFRAY, MARCHETTI ANDREWA, KRANTZ KERMITE. The Correction of Stress Incontinence by Simple Vesicourethral Suspension. J Urol 1949; 88 (4): 1326–1331. DOI: 10.1097/00005392-200210010-00005.
- &Na;. Epispadias and Incontinence. Plast Reconstr Surg 1983; 37 (5): 468. DOI: 10.1097/00006534-196605000-00023.
- Ben-Chaim J, Peppas DS, Jeffs RD, Gearhart JP. Complete Male Epispadias: Genital Reconstruction and Achieving Continence. J Urol 1995; 153 (5): 1665–1667. DOI: 10.1016/s0022-5347(01)67499-8.
- Gearhart JP, Jeffs RD. Bladder Exstrophy: Increase in Capacity Following Epispadias Repair. J Urol 1989; 142 (2 Part 2): 525–526. DOI: 10.1016/s0022-5347(17)38804-3.
- Peters CA, Gearhart JP, Jeffs RD. Epispadias and Incontinence: The Challenge of the Small Bladder. J Urol 1988; 140 (5 Part 2): 1199–1201. DOI: 10.1016/s0022-5347(17)42001-5.
- Hanna MK, Williams DI. Genital Function In Males With Vesical Exstrophy And Epispadias. Br J Urol 1972; 44 (2): 169–174. DOI: 10.1111/j.1464-410x.1972.tb10062.x.
- Klauber GT, Williams DI. Epispadias with incontinence. Plast Reconstr Surg 1974; 54 (4): 504. DOI: 10.1097/00006534-197410000-00053.
- Weiss D. Faculty Opinions recommendation of Clinical outcomes after increasing bladder outlet resistance without augmentation cystoplasty in neurogenic bladder. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2021; 17 (2): 235 1–235 7. DOI: 10.3410/f.739262243.793583985.
Dernière mise à jour: 2025-09-22 07:59