
37: Epispadias femenino
Este capítulo durará aproximadamente 16 minutos para leer.
Introducción
La extrofia vesical (BE) es una devastadora anomalía congénita del tracto urinario en la que los lactantes nacen con la vejiga urinaria exteriorizada a través de la pared abdominal, con una uretra abierta dorsalmente y con importantes anomalías secundarias de los genitales. Se considera ampliamente el trastorno congénito del tracto urinario más complejo y más desafiante desde el punto de vista quirúrgico.1 La BE varía en su grado de gravedad y existe en un continuo descrito como el complejo extrofia vesical-epispadias (BEEC).2 La incidencia del BEEC es de ~1 en 10,000.2
La extrofia cloacal (CE) se sitúa en el extremo más severo del espectro BEEC. La CE es un trastorno congénito raro con una incidencia de 1 en 200,000–400,000 nacidos vivos.3,4 Las anomalías asociadas comunes incluyen onfalocele, extrofia vesical, ano imperforado y disrafismo espinal.4 Las características adicionales incluyen una placa en la línea media de ciego extrofiado flanqueada por 2 hemivejigas, un segmento de íleon que prolapsa desde la placa cecal y un intestino posterior rudimentario de terminación ciega acompañado de un ano imperforado.4
En el extremo opuesto del espectro se encuentra el epispadias. El epispadias se presenta con mayor frecuencia como un componente de BE y CE, pero cuando el epispadias ocurre de forma aislada, en ausencia de BE y CE, se considera la variante menos grave del BEEC. El epispadias se define por una fusión incompleta de la uretra dorsal.5 Esto abarca desde un defecto glandular leve en un pene cubierto hasta la variedad penopubiana que se extiende proximalmente hasta el cuello vesical y produce incontinencia completa en varones o mujeres.5 Curiosamente, el primer caso descrito de epispadias se atribuye al emperador bizantino Heraclio (EC 610–641).6 El epispadias masculino aislado es una anomalía poco frecuente, con una incidencia reportada de 1 en 117,000 varones.6 El epispadias femenino aislado es aún más raro, con una incidencia de 1 en 484,000 pacientes de sexo femenino.6 En este capítulo nos centraremos exclusivamente en el epispadias femenino.
En 1928, Davis clasificó el epispadias femenino en tres categorías según la gravedad. Este sistema de clasificación aún se utiliza en la actualidad.7 En el grado menor de epispadias, el orificio uretral aparece simplemente pátulo. En el epispadias intermedio, la uretra se encuentra hendida dorsalmente a lo largo de la mayor parte de su longitud. En el grado más grave de epispadias, la hendidura uretral compromete toda la longitud de la uretra y el mecanismo esfinteriano queda incompetente. Cabe señalar que la prevalencia real del epispadias femenino puede ser superior a la reportada, ya que en los casos menos graves los cambios de los genitales externos pueden ser mínimos y, si no hay incontinencia, las pacientes quedan sin diagnóstico.8,9
Embriología
Una de las principales teorías del maldesarrollo embrionario en la extrofia, propuesta por Marshall y Muecke, es que el defecto básico es un sobredesarrollo anormal de la membrana cloacal durante la cuarta semana de gestación, lo que impide la migración medial del tejido mesenquimatoso y el adecuado desarrollo de la pared abdominal inferior. El momento de la rotura de esta membrana cloacal defectuosa se considera determinante de la variante resultante.10 Se piensa que existe un fracaso en el refuerzo de la membrana cloacal por el crecimiento del mesodermo. La membrana cloacal es una capa bilaminar situada en el extremo caudal del disco germinal que ocupa la pared abdominal infraumbilical. El crecimiento mesenquimatoso entre las capas ectodérmica y endodérmica de la membrana cloacal da lugar a la formación de los músculos abdominales inferiores y los huesos pélvicos. La membrana cloacal es susceptible de rotura prematura y, si esto da lugar a un pequeño defecto infraumbilical, o si la rotura ocurre en una etapa más tardía del desarrollo, se considera que se producirá epispadias, mientras que en casos de defectos mayores o de rotura más temprana, pueden dar lugar a BE o CE.10,11
Existen otras teorías plausibles sobre la causa del complejo extrofia-epispadias. Desarrollo anómalo de las eminencias genitales caudal a la posición normal, con fusión en la línea media
por debajo en lugar de por encima de la membrana cloacal, ha sido adoptada por otros investigadores.11,12 Otra hipótesis interesante que sigue siendo controvertida describe una inserción caudal anómala del pedículo corporal, lo que resulta en un fracaso de la interposición del tejido mesenquimatoso en la línea media.13 Como consecuencia de este fracaso, no se produce el desplazamiento de la cloaca hacia las profundidades de la cavidad abdominal. Una membrana cloacal que permanece en una posición infraumbilical superficial representa un estado embrionario inestable con una fuerte tendencia a desintegrarse, lo cual ha sido respaldado por el trabajo de laboratorio de Thomalla et al.14,15 Otra teoría interesante plantea que, más bien, el maldesarrollo de la pelvis ósea es el factor desencadenante del desarrollo de la extrofia. Beaudoin et al han sugerido que la falta de “rotación” del primordio del anillo pélvico impide que las estructuras adheridas al anillo pélvico se unan en la línea media, permitiendo que ocurra la herniación de la vejiga.16
Epidemiología
El epispadias femenino completo es un defecto congénito extremadamente raro, que ocurre en aproximadamente 1/484,000 nacidos vivos, mucho menos frecuente que la BE, que se estima que ocurre en 1/10,000 a 1/50,000.6 La relación hombre:mujer de la extrofia vesical derivada de múltiples series es 2.3:1 y el epispadias femenino también es menos frecuente que el epispadias masculino, que tiene una incidencia reportada de 1 en 117,000 varones.17
El riesgo de recurrencia de BE en una familia determinada es de aproximadamente 1 en 100.18 Shapiro et al en un cuestionario identificaron la recurrencia de BE y epispadias en solo 9 de aproximadamente 2,500 casos registrados.19 Shapiro et al determinaron que el riesgo de BE en la descendencia de individuos con BE y epispadias es de 1 en 70 nacidos vivos, una incidencia 500 veces mayor que en la población general.17
Patogénesis
Nuestro entendimiento actual de la patogénesis del epispadias es incompleto. Debido a la escasez de investigaciones de calidad, los expertos aún no están seguros de los orígenes embriológicos y los mecanismos genéticos que dan lugar al epispadias. Sin embargo, en los últimos años se han logrado avances respecto a la identificación de genes candidatos en el BEEC, lo que abarcaría también el desarrollo de epispadias aislado. von Lowtow et al evaluaron a 169 individuos con BEEC (reclutados en gran medida de Europa, origen genético geoétnico no informado).20 Identificaron a varios individuos con variantes patogénicas del número de copias, incluyendo uno con una duplicación previamente descrita en 22q11. Esta región también se asocia con el síndrome de deleción 22q11 (síndrome de DiGeorge), y CAKUT.21 Un metanálisis posterior de estudio de asociación del genoma completo (GWAS) que combinó 568 pacientes con BEEC y 3,241 controles de origen europeo identificó una asociación con un locus que contiene el potenciador transcripcional ISL1 (p = 2.22 × 10−08).22,23,24,25,26 Estudios funcionales y de modelos adicionales reforzaron un posible papel causal de ISL1 en el BEEC. Por ejemplo, se emplearon modelos de biología del desarrollo para aclarar la localización de la actividad de ISL1 en el tracto urinario en formación.26 El análisis de linaje genético de células que expresan ISL1 mediante un modelo murino trazador de linaje mostró células que expresan ISL1 en el tracto urinario de embriones de ratón.26
Evaluación y diagnóstico
Incluso con modalidades ecográficas modernas, el diagnóstico prenatal de la BE a menudo es difícil de delimitar, y el diagnóstico prenatal del epispadias aislado es casi imposible.27 En 2012 Goyal et al. encontraron que solo el 25% de los casos de BEEC se diagnostican prenatalmente.27 Varios grupos han intentado delinear criterios importantes para el diagnóstico de la BE de forma prenatal, pero no hay características específicas para el epispadias. Han encontrado que para la BE clásica, la ausencia de una vejiga normal llena de líquido en exploraciones repetidas sugiere el diagnóstico, al igual que una masa de tejido ecogénico en la pared abdominal inferior.28,29 Una longitud desde la inserción del cordón umbilical hasta el tubérculo genital por debajo del percentil 5 para la edad gestacional también proporciona una medida cuantitativa con la que se puede evaluar un diagnóstico de BE.30 En una revisión de 25 ecografías prenatales con el posterior nacimiento de un recién nacido con BE, Gearhart et al realizaron varias observaciones:1 ausencia de llenado vesical,2 un ombligo de implantación baja,3 ensanchamiento de la rama del pubis,4 genitales diminutos, y5 una masa en el abdomen inferior que aumentaba de tamaño a medida que avanzaba el embarazo y a medida que las vísceras intraabdominales aumentaban de tamaño.31 No es difícil entender por qué el diagnóstico prenatal del epispadias aislado es más difícil. De esas observaciones prenatales mencionadas previamente, solo un ombligo de implantación baja, una rama del pubis ensanchada y genitales diminutos podrían estar presentes potencialmente en el contexto de un epispadias aislado. Todos esos hallazgos serían menos graves en el contexto de un epispadias aislado, y los genitales diminutos no serían aplicables en el contexto de un epispadias femenino aislado.
Como resultado, el diagnóstico de epispadias femenino aislado generalmente se realiza mediante examen físico en el período posnatal. Los cambios en el aspecto externo de los genitales pueden ser mínimos, sobre todo en casos de epispadias leve. Externamente se identificará un defecto genital caracterizado por un clítoris bífido. El monte del pubis tendrá una forma deprimida y estará recubierto por una zona de piel lisa y glabra. Debajo de esta zona, puede haber una cantidad moderada de tejido subcutáneo y grasa, o la piel puede estar directamente anterior e inferior a la superficie de la sínfisis púbica. Los labios menores suelen estar poco desarrollados y terminan anteriormente en la mitad correspondiente del clítoris bífido, donde puede haber un rudimento de pliegue prepucial. La sínfisis púbica suele estar cerrada, pero puede estar separada por una estrecha banda fibrosa. Al separar los labios, se identificará la anomalía uretral, la cual puede variar considerablemente según el grado de epispadias presente. La anomalía uretral puede ir desde un defecto distal corto hasta uno que se extiende proximalmente hasta el cuello vesical (Figura 1). La vagina y los genitales internos suelen ser normales. Dado que los cambios en el aspecto externo pueden ser mínimos, algunas niñas se identifican únicamente por incontinencia persistente y, en los casos leves en los que no hay incontinencia, el diagnóstico podría no llegar a establecerse nunca. Esa es la razón por la que la verdadera prevalencia del epispadias femenino puede ser mayor que la reportada.8,9 Así, el epispadias femenino aislado siempre implica una anomalía de la uretra, pero puede haber anomalías coexistentes del cuello vesical que no deben ignorarse al manejar a estas pacientes. En formas más graves de epispadias femenino, la vejiga puede ser pequeña y comparable a la de pacientes con extrofia vesical cerrada, ya que no hubo ciclado vesical in útero. De hecho, un tercio de todos los pacientes con epispadias e incontinencia tienen una capacidad vesical menor de 60 mL y la incidencia de reflujo vesicoureteral es del 30% al 75%.32
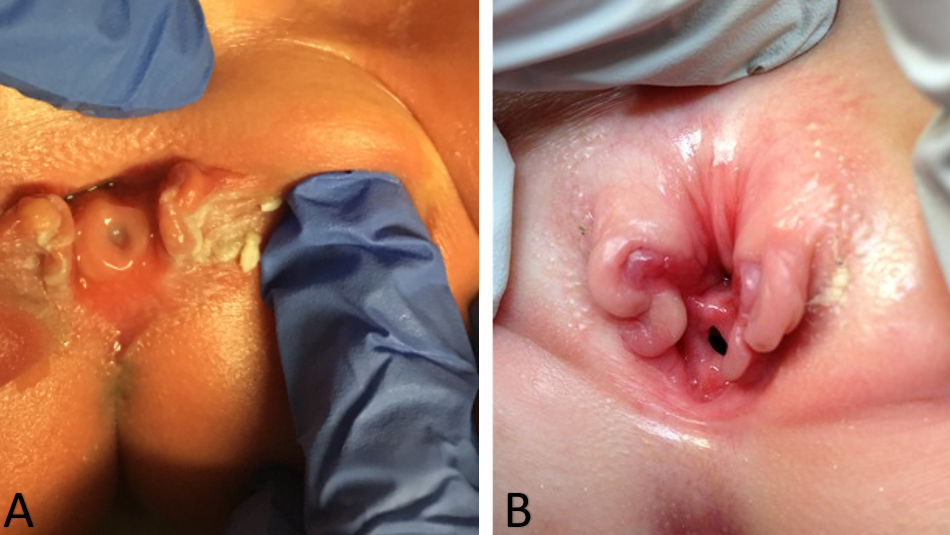
Figura 1 Casos graves de epispadias femenino. Epispadias penopúbico en una recién nacida (A). Epispadias penopúbico en una niña de 4 años (B).
Tratamiento y resultados
En la paciente con epispadias femenino, los objetivos del tratamiento son lograr la continencia urinaria, preservar las vías urinarias superiores y reconstruir unos genitales externos funcionales y estéticamente aceptables. En pacientes con epispadias leve que son continentes y presentan solo cambios mínimos en sus genitales externos, puede no ser necesaria la reconstrucción quirúrgica. En formas más graves de epispadias femenino se requiere reconstrucción quirúrgica para lograr la continencia y corregir las anomalías de los genitales externos.
Históricamente, se describieron numerosas operaciones destinadas a intentar lograr la continencia en el grupo con epispadias, pero los resultados fueron decepcionantes. Estos procedimientos incluían la plicatura transvaginal de la uretra y del cuello vesical, trasplantes musculares, torsión de la uretra, cauterización de la uretra, colgajo vesical y la suspensión vesicouretral de Marshall-Marchetti.33,34,35,36 Estos procedimientos buscaban aumentar la resistencia uretral, pero no corregían la incontinencia, ya que no intentaban normalizar la anatomía de la uretra y del cuello vesical.
Como se mencionó anteriormente, los pacientes con epispadias distal sin incontinencia pueden no requerir ninguna intervención quirúrgica. En los casos de epispadias femenino proximal, a menudo realizamos la reparación del epispadias y la reconstrucción del cuello vesical, con o sin osteotomías, al mismo tiempo (Figura 2). Esto es similar a la reparación primaria completa de la extrofia que realizamos en casos de BE y, por lo general, se lleva a cabo entre los 3–6 meses de vida. Hemos observado que la resistencia adicional obtenida con la reparación del epispadias y la reconstrucción del cuello vesical permite el ciclaje vesical y a menudo puede resultar en un aumento de la capacidad vesical. Estos hallazgos han sido corroborados por otros autores, quienes han demostrado que el cierre primario de la uretra epispádica en niños con extrofia cerrada aumenta la capacidad vesical sin causar hidronefrosis. Este enfoque se ha aplicado a pacientes masculinos y femeninos con epispadias.37,38,39
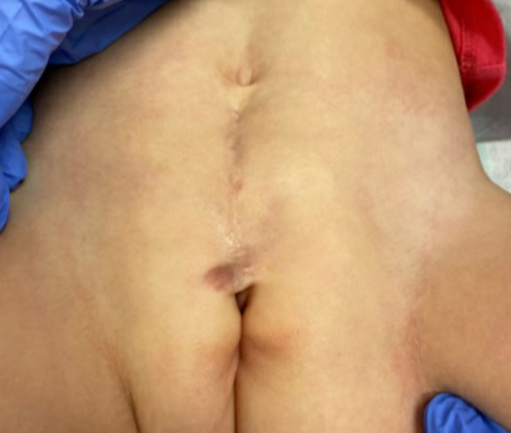
Figura 2 Imagen posoperatoria de una paciente con epispadias femenino grave, tras reparación de epispadias y reconstrucción del cuello vesical.
Las tasas de continencia en pacientes de sexo femenino con epispadias aislado se han encontrado entre 67 y 87,5 %.6,32,40 Se ha citado que el intervalo medio desde la reconstrucción hasta alcanzar la continencia es de entre 18 y 30 meses.6,41 También se han publicado informes poco frecuentes de pacientes que presentan continencia completa tardía varios años después de la reconstrucción.32 Se ha postulado que la prolongada demora en lograr la continencia es secundaria a un mayor desarrollo muscular pélvico con el tiempo y, en especial, tras los cambios hormonales de la pubertad.32
Complicaciones
La gestión quirúrgica del epispadias es notoriamente compleja y, en consecuencia, las complicaciones no son infrecuentes. Las complicaciones potenciales tras la reparación de la uretra epispádica incluyen fístula uretrocutánea, estenosis del meato uretral y estenosis uretrales. Para los pacientes que requieren un procedimiento en el cuello vesical, la importancia de un seguimiento estrecho no puede exagerarse. Cuando se incrementa la resistencia del tracto de salida vesical, los pacientes están en riesgo de desarrollar una vejiga de alta presión debido a que la vejiga se contrae para vencer la alta resistencia, lo que pone en riesgo sus vías urinarias superiores. Por ello, es fundamental que los cirujanos hagan un seguimiento muy estrecho de los pacientes tras la reconstrucción a fin de identificar precozmente la hidronefrosis de nueva aparición u otros signos de un sistema de alta presión. Esto es especialmente importante en pacientes que presentaban preoperatoriamente vejigas pequeñas y de baja complacencia, aunque esto es muy raro en el epispadias. Además, tras un procedimiento en el cuello vesical, los pacientes pueden seguir presentando incontinencia y pueden requerir procedimientos adicionales para lograr la continencia.
En pacientes con formas graves de epispadias proximal, pueden requerirse osteotomías ilíacas bilaterales en el momento de la reconstrucción urológica para disminuir la tensión del cierre al reducir la diástasis púbica. Los riesgos asociados con las osteotomías ilíacas bilaterales incluyen pérdida de sangre, disminución de la perfusión a las extremidades inferiores y, en casos muy graves, disminución de la perfusión al clítoris. La disminución de la perfusión al clítoris puede ocurrir cuando los huesos púbicos se aproximan en exceso tras las osteotomías. También son posibles fístulas vesicocutáneas en pacientes que requieren reconstrucción del cuello vesical. De manera interesante, cuando Suson et al compararon a 22 niñas con epispadias completo con 23 niñas con BE clásico, no encontraron diferencias en el número de procedimientos quirúrgicos, el desarrollo de continencia con o sin necesidad de reconstrucción del cuello vesical, ni en la eventual necesidad de derivación urinaria continente.5
Seguimiento recomendado
Los pacientes con epispadias distal que miccionan normalmente y tienen vías urinarias superiores sanas pueden, a menudo, ser simplemente vigilados y, potencialmente, dados de alta de la atención tras un período de seguimiento. En cambio, los pacientes con formas más graves de epispadias que se someten a reconstrucción deben mantenerse en seguimiento durante toda la infancia y hasta la edad adulta. Es fundamental transferir su atención, en la edad adulta, a un urólogo especializado en la atención de transición que tenga experiencia en el tratamiento de pacientes con anomalías urológicas congénitas complejas.
Como se mencionó anteriormente, los pacientes que se someten a reparación de epispadias y reconstrucción del cuello vesical requieren un seguimiento posoperatorio muy estrecho. Los estudios han demostrado que los pacientes sometidos a procedimientos aislados sobre el cuello vesical sin cistoplastia de aumento concomitante corren riesgo de desarrollar presiones vesicales elevadas y el subsiguiente deterioro del tracto urinario superior. Se ha demostrado que esto ocurre incluso cuando se realiza una evaluación preoperatoria exhaustiva para garantizar presiones y complacencia vesicales preoperatorias seguras. A pesar de la selección preoperatoria, se producen cambios significativos en la dinámica vesical y el 40% de los pacientes con vejiga neurogénica requirieron cistoplastia de aumento posterior.42 Por lo tanto, el seguimiento anual con ecografías renovesicales es absolutamente fundamental en estos pacientes.
Conclusiones
La epispadias femenina aislada es una afección sumamente rara y se considera la variante menos grave del BEEC. Varía desde un defecto distal leve que no afecta la continencia, hasta un fenotipo completo que se extiende proximalmente hasta el cuello vesical y resulta en incontinencia completa.5 Similar a la BE, el epispadias proximal se considera ampliamente uno de los trastornos congénitos del tracto urinario más complejos y de mayor desafío quirúrgico.1 En las formas más graves pueden requerirse la reparación del epispadias, la reconstrucción del cuello vesical e incluso osteotomías. Muchos pacientes con epispadias requerirán seguimiento de por vida con un urólogo que tenga experiencia en la atención de pacientes con anomalías urológicas complejas.
Puntos clave
- El epispadias es la variante menos grave del BEEC y se define por una fusión incompleta de la uretra dorsal.
- El epispadias femenino completo es un defecto congénito extremadamente raro, que ocurre en aproximadamente 1/484,000 nacidos vivos, mucho menos frecuente que la BE, cuya frecuencia se estima en 1/10,000 a 1/50,000.
- El diagnóstico del epispadias femenino aislado generalmente se realiza mediante examen físico posnatal, ya que el diagnóstico mediante imágenes prenatales no es fiable.
- La anomalía uretral puede variar desde un defecto distal corto hasta uno que se extiende proximalmente hasta el cuello vesical
- En la paciente con epispadias femenino, los objetivos del tratamiento son lograr la continencia urinaria, preservar las vías urinarias superiores y la reconstrucción de unos genitales externos funcionales y estéticamente aceptables.
- En pacientes con epispadias leve que son continentes y presentan solo cambios mínimos en sus genitales externos, puede no ser necesaria la reconstrucción quirúrgica.
- En formas más graves de epispadias femenino se requiere reconstrucción quirúrgica para obtener la continencia y corregir las anomalías de los genitales externos.
- Se ha observado que las tasas de continencia en mujeres con epispadias aislado se sitúan entre 67-87.5%.
Referencias
- Weiss DA, Shukla AR, Borer JG, Sack BS, Kryger JV, Roth EB, et al.. Evaluation of outcomes following complete primary repair of bladder exstrophy at three individual sites prior to the establishment of a multi-institutional collaborative model. J Pediatr Urol 2020; 16 (4): 435.e1–435.e6. DOI: 10.1016/j.jpurol.2020.05.153.
- Reutter H, Keppler-Noreuil K, E. Keegan C, Thiele H, Yamada G, Ludwig M. Genetics of Bladder-Exstrophy-Epispadias Complex (BEEC): Systematic Elucidation of Mendelian and Multifactorial Phenotypes. Curr Genomics 2005; 17 (1): 4–13. DOI: 10.2174/1389202916666151014221806.
- Phillips TM. Spectrum of cloacal exstrophy. Semin Pediatr Surg 2011; 20 (2): 113–118. DOI: 10.1053/j.sempedsurg.2010.12.007.
- Woo LL, Thomas JC, Brock JW. Cloacal exstrophy: A comprehensive review of an uncommon problem. J Pediatr Urol 2010; 6 (2): 102–111. DOI: 10.1016/j.jpurol.2009.09.011.
- Suson KD, Preece J, Baradaran N, Di Carlo HN, Gearhart JP. The Fate of the Complete Female Epispadias and Exstrophy Bladder–Is There a Difference? J Urol 2013; 190 (4s): 1583–1589. DOI: 10.1016/j.juro.2013.01.093.
- Gearhart JP. Exstrophy-Epispadias Complex in Campbell-Walsh-Wein Urology 2021. Elsevier; .
- Davis DM. Epispadias in Females and Its Surgical Treatment. J Urol 1928; 20 (6): 673–678. DOI: 10.1016/s0022-5347(17)73196-5.
- Yeni E, Unal D, Verit A, Karatas OF. An adult female epispadias without exstrophy was presented with urinary incontinence: a case report. Int Urogynecol J 2004; 15 (3): 212–213. DOI: 10.1007/s00192-004-1131-2.
- Krishna Shetty MV, Sen TK, Bhaskaran VA. Female epispadias. Afr J Paediatr Surg 2011; 8 (2): 215. DOI: 10.4103/0189-6725.86066.
- Muecke EC. The Role of the Cloacae Membrane in Exstrophy: The First Successful Experimental Study. J Urol 1964; 92 (6): 659–668. DOI: 10.1016/s0022-5347(17)64028-x.
- Ambrose SS, O’Brien DP. Surgical Embryology of the Exstrophy-Epispadias Complex. Surg Clin North Am 1974; 54 (6): 1379–1390. DOI: 10.1016/s0039-6109(16)40493-7.
- Patten BM, Barry A. The genesis of exstrophy of the bladder and epispadias. Am J Anat 1952; 90 (1): 35–57. DOI: 10.1002/aja.1000900103.
- Mildenberger H, Kluth D, Dziuba M. Embryology of bladder exstrophy. J Pediatr Surg 1988; 23 (2): 166–170. DOI: 10.1016/s0022-3468(88)80150-7.
- Johnston JH, Kogan SJ. The exstrophic anomalies and their surgical reconstruction. Curr Probl Surg 1974; 11 (8): 1–39. DOI: 10.1016/s0011-3840(74)80011-0.
- Thomalla JV, Rudolph RA, Rink RC, Mitchell ME. Induction of Cloacal Exstrophy in the Chick Embryo Using the CO 2 Laser. J Urol 1985; 134 (5): 991–995. DOI: 10.1016/s0022-5347(17)47573-2.
- Beaudoin S, Simon L, Bargy F. Anatomical basis of a common embryological origin for epispadias and bladder or cloacal exstrophies. Surg Radiol Anat 1997; 19 (1): 11–16. DOI: 10.1007/bf01627728.
- Shapiro E, Lepor H, Jeffs RD. The Inheritance of the Exstrophy-Epispadias Complex. J Urol 1984; 132 (2): 308–310. DOI: 10.1016/s0022-5347(17)49605-4.
- Ives E, Coffey R, Carter CO. A family study of bladder exstrophy. J Med Genet 1980; 17 (2): 139–141. DOI: 10.1136/jmg.17.2.139.
- Shapiro E, Jeffs RD, Gearhart JP, Lepor H. Muscarinic Cholinergic Receptors in Bladder Exstrophy: Insights Into Surgical Management. J Urol 1985; 134 (2): 308–310. DOI: 10.1016/s0022-5347(17)47139-4.
- Reutter H, Boyadjiev SA, Gambhir L, Ebert A-K, Rösch WH, Stein R, et al.. Phenotype Severity in the Bladder Exstrophy-Epispadias Complex: Analysis of Genetic and Nongenetic Contributing Factors in 441 Families from North America and Europe. J Pediatr 2011; 159 (5): 825–831.e1. DOI: 10.1016/j.jpeds.2011.04.042.
- WOOD HADLEYM, TROCK BRUCEJ, GEARHART JOHNP. In Vitro Fertilization and the Cloacal-Bladder Exstrophy-Epispadias Complex: Is there an Association? J Urol 2003; 169 (4): 1512–1515. DOI: 10.1097/01.ju.0000054984.76384.66.
- Lowtzow C von, Hofmann A, Zhang R, Marsch F, Ebert A-K, Rösch W, et al.. CNV analysis in 169 patients with bladder exstrophy-epispadias complex. BMC Med Genet 2016; 17 (1): 35. DOI: 10.1186/s12881-016-0299-x.
- Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, et al.. Copy-Number Disorders Are a Common Cause of Congenital Kidney Malformations. Am J Hum Genet 2012; 91 (6): 987–997. DOI: 10.1016/j.ajhg.2012.10.007.
- Arkani S, Cao J, Lundin J, Nilsson D, Källman T, Barker G, et al.. Evaluation of the ISL1 gene in the pathogenesis of bladder exstrophy in a Swedish cohort. Hum Genome Var 2018; 5 (1): 18009. DOI: 10.1038/hgv.2018.9.
- Draaken M, Knapp M, Pennimpede T, Schmidt JM, Ebert A-K, Rösch W, et al.. Genome-wide Association Study and Meta-Analysis Identify ISL1 as Genome-wide Significant Susceptibility Gene for Bladder Exstrophy. PLoS Genet 2015; 11 (3): e1005024. DOI: 10.1371/journal.pgen.1005024.
- Reutter H, Draaken M, Pennimpede T, Wittler L, Brockschmidt FF, Ebert A-K, et al.. Genome-wide association study and mouse expression data identify a highly conserved 32 kb intergenic region between WNT3 and WNT9b as possible susceptibility locus for isolated classic exstrophy of the bladder. Hum Mol Genet 2014; 23 (20): 5536–5544. DOI: 10.1093/hmg/ddu259.
- Reutter H, Hoischen A, Ludwig M, Stein R, Radlwimmer B, Engels H, et al.. Genome-wide analysis for micro-aberrations in familial exstrophy of the bladder using array-based comparative genomic hybridization. BJU Int 2007; 100 (3): 646–650. DOI: 10.1111/j.1464-410x.2007.07086.x.
- Zhang R, Knapp M, Suzuki K, Kajioka D, Schmidt JM, Winkler J, et al.. ISL1 is a major susceptibility gene for classic bladder exstrophy and a regulator of urinary tract development. Sci Rep 2017; 7 (1): 42170. DOI: 10.1038/srep42170.
- Goyal A, Fishwick J, Hurrell R, Cervellione RM, Dickson AP. Antenatal diagnosis of bladder/cloacal exstrophy: Challenges and possible solutions. J Pediatr Urol 2012; 8 (2): 140–144. DOI: 10.1016/j.jpurol.2011.05.003.
- Mirk P, Calisti A, Fileni A. Prenatal sonographic diagnosis of bladder extrophy. J Ultrasound Med 1986; 5 (5): 291–293. DOI: 10.7863/jum.1986.5.5.291.
- VERCO PW, KHOR BH, BARBARY J, ENTHOVEN C. Ectopia Vesicae in Utero. Australas Radiol 1986; 30 (2): 117–120. DOI: 10.1111/j.1440-1673.1986.tb02400.x.
- Fishel-Bartal M, Perlman S, Messing B, Bardin R, Kivilevitch Z, Achiron R, et al.. Early Diagnosis of Bladder Exstrophy: Quantitative Assessment of a Low-Inserted Umbilical Cord. J Ultrasound Med 2017; 36 (9): 1801–1805. DOI: 10.1002/jum.14212.
- GEARHART J, BENCHAIM J, JEFFS R, SANDERS R. Criteria for the prenatal diagnosis of classic bladder exstrophy. Obstet Gynecol 1995; 85 (6): 961–964. DOI: 10.1016/0029-7844(95)00069-4.
- Kelalis PP, Kramer SA. Surgical Correction of Female Epispadias. Eur Urol 1982; 8 (6): 321–324. DOI: 10.1159/000473547.
- Jonuzi A, Popović N, Zvizdić Z, Milišić E, Karavdić K, Dewan P. Female Epispadias Presenting as Urinary Incontinence. APSP J Case Rep 2017; 8 (2): 10. DOI: 10.21699/ajcr.v8i2.548.
- Gross RE, Cresson SL. Exstrophy Of Bladder. J Am Med Assoc 1952; 149 (18): 1640. DOI: 10.1001/jama.1952.02930350028008.
- MARSHALL VICTORFRAY, MARCHETTI ANDREWA, KRANTZ KERMITE. The Correction of Stress Incontinence by Simple Vesicourethral Suspension. J Urol 1949; 88 (4): 1326–1331. DOI: 10.1097/00005392-200210010-00005.
- &Na;. Epispadias and Incontinence. Plast Reconstr Surg 1983; 37 (5): 468. DOI: 10.1097/00006534-196605000-00023.
- Ben-Chaim J, Peppas DS, Jeffs RD, Gearhart JP. Complete Male Epispadias: Genital Reconstruction and Achieving Continence. J Urol 1995; 153 (5): 1665–1667. DOI: 10.1016/s0022-5347(01)67499-8.
- Gearhart JP, Jeffs RD. Bladder Exstrophy: Increase in Capacity Following Epispadias Repair. J Urol 1989; 142 (2 Part 2): 525–526. DOI: 10.1016/s0022-5347(17)38804-3.
- Peters CA, Gearhart JP, Jeffs RD. Epispadias and Incontinence: The Challenge of the Small Bladder. J Urol 1988; 140 (5 Part 2): 1199–1201. DOI: 10.1016/s0022-5347(17)42001-5.
- Hanna MK, Williams DI. Genital Function In Males With Vesical Exstrophy And Epispadias. Br J Urol 1972; 44 (2): 169–174. DOI: 10.1111/j.1464-410x.1972.tb10062.x.
- Klauber GT, Williams DI. Epispadias with incontinence. Plast Reconstr Surg 1974; 54 (4): 504. DOI: 10.1097/00006534-197410000-00053.
- Weiss D. Faculty Opinions recommendation of Clinical outcomes after increasing bladder outlet resistance without augmentation cystoplasty in neurogenic bladder. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2021; 17 (2): 235 1–235 7. DOI: 10.3410/f.739262243.793583985.
Última actualización: 2025-09-21 13:35