
27: Syndrome prune-belly
Ce chapitre prendra environ 30 minutes de lecture.
Introduction
Le syndrome de prune-belly (PBS), également appelé syndrome d’Eagle-Barrett, est une affection multisystémique rare généralement caractérisée par une constellation d’anomalies de sévérité variable, dont les principales sont un déficit de la musculature abdominale, des testicules intra-abdominaux bilatéraux et des anomalies des voies urinaires.1,2Des anomalies associées supplémentaires non génito-urinaires touchent l’appareil respiratoire, l’appareil gastro-intestinal, le système cardiaque et le système musculo-squelettique.3 La sévérité de la maladie s’inscrit sur un large continuum, certains enfants ne survivant pas à la période néonatale et d’autres étant peu atteints.4
Embryologie
Plusieurs théories prédominantes ont été proposées concernant l’embryogenèse du PBS, bien qu’aucune ne soit universellement acceptée, et il existe un certain chevauchement entre elles. Les quatre principales théories sont 1, une obstruction urétrale postérieure précoce in utero (probablement une prostate hypoplasique/dysplasique ou un urètre anormal), entraînant une dilatation proximale de la vessie, des uretères et des reins, associée à un développement insuffisant de la paroi abdominale 2, une anomalie primaire du mésoderme de la lame latérale, qui est le précurseur des uretères, de la vessie, de la prostate, de l’urètre et du gouvernaculum 3, une anomalie intrinsèque des voies urinaires conduisant à une dilatation urétérale et à une ascite fœtale et 4, une anomalie du sac vitellin.5,6
Épidémiologie
L’incidence du PBS a été estimée entre 1 sur 29 000 et 1 sur 40 000 naissances vivantes, similaire à celle de l’exstrophie vésicale, 95 % des patients atteints étant de sexe masculin.1,2,7 Les filles représentent moins de 5 % de tous les cas de PBS et présentent une déficience de la paroi abdominale et un tractus urinaire dilaté et dysmorphique, sans anomalie gonadique associée.8 Une incidence plus élevée est observée chez les jumeaux, les personnes noires et les enfants nés de mères plus jeunes.9 Bien que le PBS se présente souvent comme un événement sporadique, une contribution génétique est également suspectée, avec des mutations dans des gènes codant des facteurs de transcription du muscle lisse, des filaments contractiles et des enzymes/récepteurs neuronaux.10,11
Caractéristiques cliniques et physiopathologie
Anomalies génito-urinaires
Reins
Le spectre des anomalies rénales chez les patients atteints de PBS s’étend d’un parenchyme rénal normal à la dysplasie. Environ la moitié des cas présentent une dysplasie rénale, le plus souvent des variétés de type Potter II et IV, et cela peut également varier entre les deux côtés.1,2 Alors que la variété de type II, avec peu de néphrons et une désorganisation parenchymateuse, est plus évocatrice d’un défaut mésenchymateux rénal, la variété de type IV, avec des kystes corticaux et tubulaires, est associée à une obstruction de la voie d’évacuation, en l’absence de décompression ouraquienne.12 Bien que le système collecteur rénal soit caractéristiquement dilaté, souvent de façon sévère, le degré de dilatation n’est toutefois pas corrélé au degré de dysplasie rénale et la morphologie calicielle peut être bien préservée, même en présence d’une dilatation massive. Bien que l’hydronéphrose soit habituellement non obstructive, une obstruction de la jonction pyélo-urétérale primaire ou secondaire peut également survenir. Cependant, il est important de noter que c’est l’infection rénale, plutôt que l’obstruction, qui constitue le risque le plus important pour la fonction rénale chez ces patients.1,2
Uretères
Les uretères sont généralement dilatés, tortueux et redondants. Bien qu’une dilatation massive et une sténose puissent survenir à tous les niveaux, les portions proximales (supérieures) des uretères sont habituellement moins anormales que les segments distaux. Le reflux vésico-urétéral (RVU) est présent chez 75 % des enfants atteints de PBS.12 L’obstruction n’est pas fréquente mais a été rapportée aux jonctions pyélo-urétérale et urétéro-vésicale. Les coupes histologiques montrent un déficit en cellules musculaires lisses et une augmentation du rapport collagène/muscle lisse dans les segments distaux les plus atteints, en particulier ceux qui présentent un reflux, avec des cellules musculaires lisses d’aspect plus normal dans les segments proximaux.13 Ce fait est crucial lorsque l’on entreprend une reconstruction urétérale. La diminution du nombre de myofibrilles épaisses et fines observée à l’examen ultrastructural est considérée comme contribuant à un péristaltisme inefficace en raison d’une mauvaise coaptation de la paroi urétérale, entraînant une stase des voies urinaires supérieures susceptible de conduire à une infection. En outre, chez de nombreux patients, la gravité des anomalies des voies urinaires n’est pas proportionnelle au degré de flaccidité de la paroi abdominale.14
Vessie
La vessie apparaît généralement massivement distendue, avec un pseudo-diverticule de l’ouraque et un ouraque perméable chez 25 % à 30 % des patients.12 Contrairement à l’aspect observé dans les vessies obstruées pour d’autres raisons , malgré sa grande épaisseur, le contour vésical est régulier. À la miction, le col vésical s’ouvre largement dans un urètre prostatique dilaté.1,2 Histologiquement, en l’absence d’obstruction, la vessie présente un rapport collagène/fibres musculaires augmenté ; cependant, une hypertrophie du muscle lisse peut être observée en cas d’obstruction. En cystoscopie , le trigone est évasé avec des méats urétéraux déplacés vers le haut et latéralement , ce qui peut contribuer à la forte incidence du reflux vésico-urétéral. Parmi les constatations caractéristiques d’un bilan urodynamique figurent une vessie de compliance normale avec retard de la première sensation de besoin de miction et une grande capacité.15 L’efficacité mictionnelle de ces vessies est variable, certaines se vidant complètement tandis que d’autres présentent un résidu post-mictionnel important, probablement dû à une obstruction cervico-urétrale relative et à l’incapacité du détrusor à générer une pression suffisante pendant la miction chez ces patients.16 Le terme « miction déséquilibrée » est généralement utilisé pour désigner la première situation où une résistance relative à l’écoulement empêche une vidange vésicale efficace.1,2 Bien que 50 % des patients atteints du syndrome prune-belly mictionnent spontanément avec des pressions mictionnelles normales, des débits normaux et de faibles résidus post-mictionnels, il a été observé qu’une détérioration de la miction équilibrée peut survenir avec la croissance, entraînant des résidus post-mictionnels significatifs, ce qui souligne la nécessité d’une évaluation périodique.16
Prostate et organes génitaux accessoires
La dilatation urétrale postérieure est due à une hypoplasie prostatique, probablement liée à un développement mésenchymo-épithélial anormal.12,13,14,15,16,17,18,19 Les coupes histologiques révèlent peu d’éléments cellulaires prostatiques, avec une diminution des cellules épithéliales et des cellules musculaires lisses et une augmentation des cellules du tissu conjonctif.19 Environ 20 % des patients présentent également des lésions obstructives de l’urètre postérieur distal sous la forme d’une atrésie urétrale, de valves, d’une sténose urétrale, d’une membrane urétrale et d’un diverticule urétral. L’absence de tissu parenchymateux prostatique peut entraîner une angulation de l’urètre lors de la miction, désignée par Stephens sous le terme de valve de type IV.12 L’hypoplasie prostatique et l’incompétence associée du col vésical sont également considérées comme un facteur d’échec de l’éjaculation/ éjaculation rétrograde chez les patients atteints de PBS.1,2 Les organes génitaux accessoires tels que le canal déférent et les vésicules séminales sont souvent atrétiques, bien que l’un ou l’autre puisse être dilaté ou épaissi. L’épididyme peut être faiblement attaché au testicule et il peut également exister une absence de continuité entre les canalicules efférents et le rete testis.20
Urètre antérieur
Bien que l’urètre antérieur de l’enfant atteint de PBS soit généralement normal, plusieurs anomalies du segment urétral ont été rapportées; les plus fréquentes sont l’atrésie ou l’hypoplasie urétrale et le mégalourètre.21,22,23,24,25 Il a été postulé que l’atrésie ou l’hypoplasie urétrale survient parce que l’urètre n’est pas utilisé, plutôt que malformé. L’atrésie urétrale est souvent létale si elle n’est pas associée à une décompression sous forme d’un ouraque perméable ou parfois même d’une rupture vésicale spontanée avec formation de fistule.22
Une caractéristique unique du PBS est la survenue d’un méga-urètre.26 Une obstruction transitoire in utero de la jonction entre l’urètre glandulaire et l’urètre pénien a été proposée comme cause du méga-urètre. Deux variantes de méga-urètre sont observées chez les patients atteints de PBS — à savoir la forme fusiforme (Figure 1) et la forme scaphoïde (Figure 2), (Figure 3).24 La forme fusiforme correspond à une déficience des corps caverneux ainsi que du corps spongieux, probablement résultant d’une insuffisance mésenchymateuse des replis urétraux, et se manifeste cliniquement par une dilatation de l’ensemble du phallus pendant la miction. À l’inverse, la forme scaphoïde, qui découlerait probablement d’une insuffisance mésenchymateuse des tissus de soutien de l’urètre entraînant une déficience limitée au seul corps spongieux avec préservation du gland et des corps caverneux, se caractérise par une dilatation de l’urètre ventral pendant la miction.26
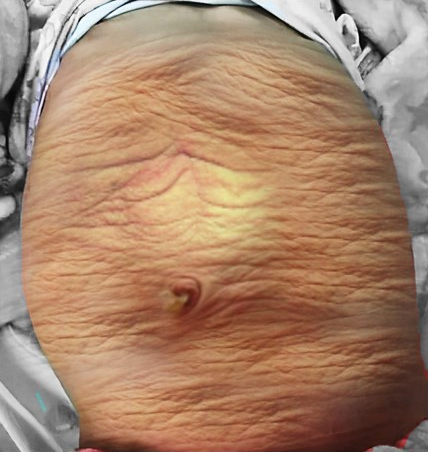
Figure 1 Photographie clinique d’un enfant atteint du syndrome de l’abdomen en prune
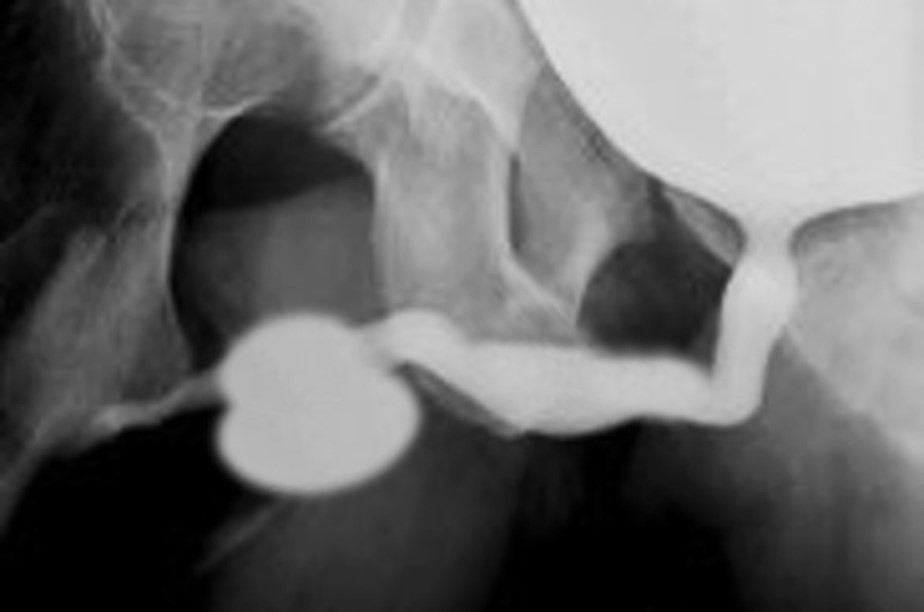
Figure 2 Méga-urètre fusiforme
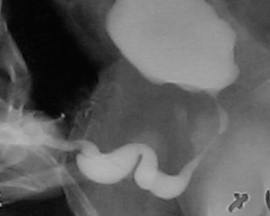
Figure 3 Méga-urètre scaphoïde
Testicules
Des testicules intra-abdominaux bilatéraux situés au-dessus des vaisseaux iliaques et adjacents aux uretères dilatés sont considérés comme l’une des constatations typiques du PBS.1,2 Bien que des forces mécaniques, telles qu’une obstruction mécanique due à une mégavessie et une faible pression intra-abdominale liée à des anomalies de la paroi abdominale, aient longtemps été avancées comme l’étiologie de cet échec de la descente testiculaire, le fait que certains patients présentant les anomalies typiques des voies urinaires et de la musculature abdominale puissent avoir des testicules descendus jette un doute sur la responsabilité exclusive de facteurs purement mécaniques.1,2 S’agissant de savoir si les testicules des enfants atteints de PBS sont intrinsèquement différents de ceux sans PBS, Pak et al ont comparé l’histologie de testicules intra-abdominaux et n’ont trouvé aucune différence dans le nombre de cellules germinales, de spermatogonies et de cellules de Leydig entre les testicules PBS et les testicules fœtaux intra-abdominaux non PBS.27,28 Cependant, Orvis et al ont noté une diminution du nombre de spermatogonies et une hyperplasie des cellules de Leydig dans des testicules fœtaux PBS, ce qui implique une anomalie testiculaire intrinsèque.29 Concernant le risque de malignité dans ces testicules, bien que ce risque puisse être relativement faible en raison de l’absence d’épithélium germinal, une orchidopexie précoce et un suivi au long cours sont nécessaires pour réduire le risque de malignité testiculaire et en améliorer la détection.30,31
Défaut de la paroi abdominale
L’apparence de la paroi abdominale est une caractéristique chez les enfants atteints de PBS. Le plus souvent, la déficience musculaire est inégale et par plaques, les segments musculaires médiaux et inférieurs étant généralement les plus déficients, et elle peut être disproportionnée par rapport aux anomalies des voies urinaires. Les zones les plus sévèrement atteintes peuvent comporter de la peau, de la graisse sous-cutanée et une couche fibreuse unique sur le péritoine, l’examen histologique au microscope électronique montrant un aspect non spécifique de désorganisation des myofilaments, une désorganisation des lignes Z et une prolifération mitochondriale.32,33 Malgré ces problèmes de paroi abdominale, on observe chez ces enfants une bonne cicatrisation sans tendance aux infections ni aux hernies incisionnelles. L’aspect typique à la naissance est celui d’une peau plissée et redondante, avec un abdomen qui fait saillie aux flancs. À mesure que les patients grandissent, certains montrent une amélioration du tonus et de l’apparence abdominaux, avec moins de plis, tandis que chez d’autres l’abdomen prend davantage un aspect de ventre en pot.1,2 La démarche n’est généralement pas affectée, bien qu’elle puisse être retardée, et les enfants ont tendance à se tourner sur le côté et à utiliser leurs bras pour s’asseoir à partir de la position de décubitus dorsal. Le faible soutien de la paroi thoracique inférieure peut entraîner un évasement de la marge costale et, l’efficacité de la toux s’en trouvant compromise, rendre ces enfants plus vulnérables aux affections respiratoires.
Anomalies extra-génito-urinaires
Parmi tous les enfants atteints de PBS, 75 % présentent des anomalies extra-urinaires.1,2,4 Après le défaut évident de la paroi abdominale, les problèmes les plus fréquents sont cardiaques, pulmonaires et orthopédiques (Tableau 1).34,35,36,37,38,39,40 Outre ces morbidités spécifiques d’organe, près de 50 % des enfants atteints de PBS naissent prématurés, ce qui contribue de manière significative aux comorbidités.
Tableau 1 Anomalies extra-génito-urinaires fréquentes observées dans le syndrome de prune-belly
| Système | Anomalies fréquentes | Remarques |
|---|---|---|
| Anomalies cardiaques (10%) | Canal artériel persistant, communications interauriculaire et interventriculaire, communication interventriculaire, et tétralogie de Fallot | Les anomalies cardiaques à la naissance peuvent primer sur les problèmes urologiques. |
| Anomalies pulmonaires (55%) | Hypoplasie pulmonaire Pneumothorax et pneumomédiastin peuvent être observés avec ou sans hypoplasie pulmonaire | L’hypoplasie pulmonaire peut résulter d’un oligohydramnios sévère lié à une dysplasie rénale ou à une obstruction sévère de la voie d’évacuation vésicale et peut entraîner le décès du nouveau-né. Chez près de la moitié des nouveau-nés atteints de PBS, une intubation et une ventilation mécanique seront nécessaires, avec les morbidités qui y sont associées. L’incapacité à générer une pression intra-abdominale significative et les anomalies musculosquelettiques associées telles que la scoliose et les anomalies de la cage thoracique, le plus souvent un pectus excavatum, peuvent contribuer à un risque accru de pneumonie et d’atélectasie lobaire chez ces enfants. |
| Anomalies gastro-intestinales (30%) | Les anomalies résultent d’une rotation incomplète de l’intestin moyen entraînant un mésentère large, ce qui accroît la mobilité intestinale avec malrotation intestinale, volvulus, atrésies et sténoses. Une torsion splénique liée à une fixation mésentérique anormale a également été rapportée. Omphalocèle, gastroschisis, situs inversus abdominis, VACTERL (défauts vertébraux, atrésie anale, malformations cardiaques, fistule trachéo-œsophagienne, anomalies rénales et anomalies des membres) et anomalies anorectales ont été signalées. | Du fait de la capacité limitée à générer une pression intra-abdominale en raison de l’hypoplasie des muscles abdominaux, la constipation devient un problème à vie et peut conduire à un mégacôlon acquis. |
| Anomalies orthopédiques (65%) | La présence de fossettes sur la face latérale des genoux est une constatation fréquente en cas d’oligohydramnios et elle est fréquemment observée chez les patients atteints de PBS, avec d’autres anomalies telles que le pied bot varus équin (26 %), la dysplasie de la hanche (5 %), et la scoliose congénitale (4 %). Hypoplasie, absence ou amputation des membres inférieurs. | Compte tenu du fait que la plupart de ces anomalies musculosquelettiques sont unilatérales, la cause la plus probable de ces anomalies est l’effet compressif de l’oligohydramnios. Un autre mécanisme proposé pour les anomalies des membres inférieurs est la compression des vaisseaux iliaques externes par la vessie distendue, compromettant ainsi l’apport sanguin aux membres inférieurs. De plus, certaines de ces malformations, telles que l’ostéodystrophie rénale, la luxation de la hanche, la scoliose et le pectus excavatum ou carinatum, ont tendance à se manifester ou à s’aggraver avec la croissance. |
Diagnostic et évaluation
Prénatal
Bien que le diagnostic de PBS à l’échographie ait été rapporté dès 11-12 semaines de grossesse, la précision diagnostique varie de 30 % à 85 % car le PBS se présente en période prénatale avec de nombreux signes échographiques comparables à ceux d’une obstruction des voies urinaires basses (LUTO) due à d’autres causes, notamment des valves urétrales postérieures, une urétérocèle et une atrésie urétrale.40,41,42,43,44 Le diagnostic prénatal de PBS doit être envisagé chaque fois que les anomalies échographiques suivantes sont clairement identifiées : oligoamnios, anomalies urinaires (dilatation des voies urinaires, mégavessie, hydro-urétéro-néphrose bilatérale) et absence de musculature abdominale. Un diagnostic précoce améliore non seulement la survie en incitant le clinicien à organiser l’accouchement dans un centre tertiaire pour une prise en charge multidisciplinaire rapide des nouveau-nés atteints, mais offre également aux futurs parents, s’ils le souhaitent, la possibilité d’une interruption volontaire de grossesse.43
Nouveau-né
L’aspect de la paroi abdominale est diagnostique chez le nouveau-né, indépendamment du fait que le diagnostic ait été connu en période prénatale. Les enfants atteints de PBS posent un défi unique et nécessitent une prise en charge multidisciplinaire rapide par une équipe comprenant un néonatologue, un urologue, un néphrologue, ainsi qu’un cardiologue et un orthopédiste pédiatrique lorsque cela est indiqué. Étant donné la grande variabilité de la sévérité de la maladie, une évaluation multisystémique visant à exclure des malformations cardiaques, pulmonaires ou autres associées significatives doit avoir lieu en période néonatale. Une radiographie thoracique immédiate est nécessaire pour exclure un pneumothorax et un pneumomédiastin associés. Comme c’est le cas avec la LUTO, l’évolution postnatale initiale est dictée par la gravité des comorbidités telles que l’hypoplasie pulmonaire, avec des issues plus défavorables en cas de prématurité et d’hypoplasie pulmonaire sévère.45,46,47
L’oligoamnios anténatal chez ces nourrissons indique une altération de la fonction rénale. Les dosages initiaux de créatinine reflètent la fonction rénale maternelle et des prélèvements répétés sont nécessaires. Si, après 48–72 heures, la créatinine sérique est supérieure à 1,0 chez le nouveau-né à terme ou à 1,5 chez le nouveau-né prématuré, un degré d’insuffisance rénale est présent. Une augmentation progressive de la créatinine sérique au cours des premières semaines de vie annonce un pronostic défavorable. Si le nadir initial de la créatinine est inférieur à 0,7 mg/dL, une insuffisance rénale ultérieure est peu probable.48 Les urines sont envoyées pour une culture initiale et une prophylaxie antibiotique est instaurée. La mesure des électrolytes sériques et urinaires à la naissance peut être utile pour apprécier l’épargne sodée, témoignant d’une fonction rénale adéquate.
Évaluation diagnostique
Une évaluation urologique approfondie est réalisée après la stabilisation du patient compte tenu des autres comorbidités. Un examen clinique et une échographie des voies urinaires sont initialement requis.46 Les échographies rénales fournissent des informations sur l’épaisseur corticale, la présence de remaniements kystiques et la taille rénale. Les examens échographiques avant et après la miction donnent une indication de la présence d’un reflux vésico-urétéral et d’un résidu urinaire post-mictionnel.
Initialement, une cysto-urétrographie mictionnelle (VCUG) est réalisée chez les nourrissons présentant une fonction rénale anormale afin de s’assurer qu’il n’existe pas de véritable obstruction urétrale anatomique. Cette “variante létale” et la confusion potentielle avec des valves urétrales postérieures “se faisant passer” pour un syndrome prune-belly doivent être diagnostiquées, car le traitement change radicalement. De plus, la VCUG met en évidence un reflux vésico-urétéral chez jusqu’à 85% des patients et évalue la vidange vésicale. Tous les nourrissons sont traités par antibiotiques avant un cathétérisme stérile afin de diminuer le risque d’infection. En général, l’évaluation radiologique de l’appareil urinaire est adaptée à la philosophie de prise en charge de chaque chirurgien urologue. Les examens avec contraste nécessitant une instrumentation à risque sont évités chez les nourrissons ayant une bonne fonction rénale.22
Après l’évaluation initiale, une scintigraphie rénale à la mercaptoacétyl‑triglycine (MAG3) peut fournir des informations à la fois fonctionnelles et anatomiques. Le flux sanguin, la fonction rénale différentielle et le drainage en réponse au furosémide (Lasix) peuvent être évalués et comparés aux examens ultérieurs. Les limites de la rénographie de diurèse pour démontrer une obstruction chez les patients atteints du syndrome du ventre en prune ont été notées.49 Un test de Whitaker est parfois nécessaire en tant qu’examen de référence pour démontrer une obstruction significative, bien que cela soit principalement d’intérêt historique. Une scintigraphie rénale à l’acide dimercaptosuccinique (DMSA) est réalisée plutôt qu’une urographie excrétrice (IVP) pour évaluer la taille rénale, la masse tubulaire et la fonction différentielle. Les études comparatives sont inestimables pour évaluer la croissance rénale et les cicatrices, car ces patients nécessitent un suivi à long terme. Certains groupes ont préconisé l’urographie par résonance magnétique (MRU) chez ces patients en raison de sa haute résolution et de ses excellentes données fonctionnelles et anatomiques, qui permettent une visualisation détaillée des anomalies des voies urinaires supérieures et peuvent également être utiles pour la planification opératoire.50 Comme indiqué dans Tableau 1, il existe trois grandes catégories de présentation néonatale telles que décrites par Woodard.1,2
Prise en charge et résultats
Prise en charge prénatale
Intervention in utero
Avec l’amélioration de l’imagerie materno-fœtale avancée et des techniques diagnostiques, le syndrome du prune-belly peut être suspecté à l’imagerie dès le début de la grossesse. L’intervention fœtale traite habituellement l’oligoamnios ou l’anhydramnios associés à l’hypoplasie pulmonaire, principale cause de mortalité périnatale. Les critères d’une intervention intra-utérine comprennent une gestation au deuxième ou au troisième trimestre, un oligoamnios sévère, une mégacystis, une hydronéphrose avancée, un caryotype normal, une étendue limitée d’autres anomalies congénitales détectées et des analyses urinaires fœtales aspirées sériées favorables.51,52,53 Bien que plusieurs rapports de procédures de shunt vésico-amniotique pour uropathie obstructive fœtale existent, les revues n’ont pas réussi à documenter un effet bénéfique de l’intervention fœtale sur la fonction rénale ultérieure et la fonction pulmonaire ne peut pas davantage être assurée malgré la restauration de niveaux normaux de liquide amniotique. Comme la majorité des patients atteints du syndrome du prune-belly ne présentent aucune preuve démontrable d’obstruction et que la fonction rénale ne corrèle pas avec le degré de dilatation présent dans les voies urinaires, les risques et les bénéfices d’une intervention in utero doivent être soigneusement pesés.53
Prise en charge postnatale
Les objectifs globaux de la prise en charge sont de préserver la fonction rénale et de prévenir les infections. La réalisation de ces objectifs est possible grâce à diverses approches thérapeutiques, allant de l’abstention surveillée à la reconstruction chirurgicale immédiate des voies urinaires. Une intervention chirurgicale précoce chez le nouveau-né est évitée, sauf en cas d’élévation de la créatinine ou d’infection, nécessitant une vésicostomie précoce. Comme indiqué dans Tableau 2, la prise en charge de ces patients dépend de la catégorie de sévérité à la présentation.
Tableau 2 Présentation et prise en charge des différentes catégories basées sur les critères de présentation néonatale de Woodward
| Catégorie de gravité | Présentation | Prise en charge | Suivi et résultats |
|---|---|---|---|
| I | Oligoamnios marqué secondaire à une dysplasie rénale et/ou à une obstruction de la voie urinaire d’évacuation, entraînant une hypoplasie pulmonaire sévère et des anomalies squelettiques | Chez les rares nourrissons survivant à l’atteinte pulmonaire, une dérivation urinaire haute par pyélostomie cutanée a été tentée; toutefois, la récupération de la fonction rénale n’est généralement pas possible en raison d’une dysplasie rénale sous-jacente sévère. | Ces nouveau-nés décèdent souvent en quelques jours et les interventions sont limitées. Associés à un oligoamnios prénatal et à une hypoplasie pulmonaire postnatale, ils meurent habituellement en période postnatale immédiate en raison de complications pulmonaires. |
| Un faciès de Potter peut être présent et est secondaire à l’oligoamnios. Les caractéristiques comprennent une micrognathie, des yeux largement écartés, des fentes palpébrales aplaties, un épicanthus marqué, une arête nasale aplatie, des oreilles basses dépourvues de cartilage et des déformations squelettiques. | Seul un cathétérisme peut être nécessaire | ||
| Les cas d’atrésie urétrale relèvent typiquement de cette catégorie la plus sévère. | |||
| II | Insuffisance rénale modérée et hydrouréteronéphrose modérée à sévère. L’hypoplasie pulmonaire n’est pas une caractéristique majeure. | Ces enfants reçoivent des antibiotiques prophylactiques au long cours et sont surveillés de façon sériée pour dépister une infection et toute diminution de la fonction rénale. | Une surveillance postopératoire à vie est nécessaire pour évaluer la fonction rénale et le drainage optimal des voies urinaires supérieures et inférieures au moyen d’études fonctionnelles et anatomiques. |
| Une reconstruction agressive des voies urinaires est envisagée en cas de documentation d’une infection, d’absence de croissance rénale adéquate ou de diminution de la fonction rénale. | |||
| Cependant, la reconstruction chirurgicale est habituellement différée jusqu’à l’âge de 3 mois ou plus afin de permettre la maturation pulmonaire. | |||
| Si l’insuffisance rénale est sévère ou si une infection est présente d’emblée, une période de dérivation urinaire par vésicostomie cutanée est recommandée. | |||
| Les urétérostomies cutanées apportent probablement peu pour améliorer le drainage. De plus, l’uretère proximal est anatomiquement et histologiquement le plus normal et devrait être préservé si une reconstruction adaptée est envisagée ultérieurement. | |||
| III | Manifestations légères de la triade ou formes incomplètes | Constituent la majorité des patients et présentent une bonne fonction rénale, malgré une dilatation marquée des voies urinaires supérieures et inférieures. | Comme pour la catégorie 2, une surveillance à vie est nécessaire. |
| La fonction rénale est généralement normale ou légèrement altérée et il n’y a pas d’insuffisance pulmonaire. | Une intervention chirurgicale précoce de reconstruction des voies urinaires n’est généralement pas nécessaire, et ces patients sont pris en charge par une prophylaxie antibiotique au long cours et une évaluation sériée de la fonction rénale à l’aide de techniques d’imagerie sélective. | La survenue d’infections urinaires ou une diminution de la fonction rénale impose une évaluation spécifique complémentaire sous la forme d’un bilan urodynamique ou d’un rénogramme diurétique afin de documenter une obstruction des voies basses ou hautes pouvant nécessiter une intervention. | |
| Ces patients bénéficient d’une orchidopéxie et d’une abdominoplastie précoces, la correction de la paroi abdominale pouvant améliorer l’efficacité de la défécation et de la miction. |
Interventions chirurgicales dans le syndrome de prune-belly
Voies urinaires
Une constatation caractéristique du PBS est une dilatation à basse pression de l’ensemble des voies urinaires, s’étendant du bassinet rénal en amont jusqu’à l’urètre en aval. La vessie est généralement augmentée de volume et hypotonique, avec une compliance élevée et un reflux vésico-urétéral (VUR) à basse pression présent chez environ 75 % des patients. Bien que ces vessies de grande capacité et à haute compliance permettent un stockage à basse pression, elles présentent souvent une vidange incomplète secondaire à une contractilité détrusorienne réduite. Par conséquent, l’intervention urologique initiale est dirigée vers le drainage vésical afin d’éviter l’infection urinaire (IU) et de préserver ainsi la fonction rénale. À ce titre, la circoncision, les antibiotiques prophylactiques et la miction programmée/double miction sont souvent mis en œuvre pour diminuer l’incidence des IU et protéger les voies urinaires supérieures de nouvelles agressions.54,55,56,57 Lorsque cette prise en charge conservatrice ne parvient pas à prévenir l’infection et à obtenir une vidange adéquate, ces enfants peuvent nécessiter une cystoplastie de réduction et/ou une appendicovésicostomie afin de faciliter le cathétérisme intermittent propre et/ou une chirurgie antireflux pour la prise en charge du VUR associé.58,59,60
Paroi abdominale
La reconstruction de la paroi abdominale, où la paroi abdominale périphérique plus normale est avancée pour soutenir cette portion centrale anormale et déficiente, est un élément important de la prise en charge chirurgicale des patients atteints de PBS. Outre l’esthétique, il a également été démontré que l’abdominoplastie améliore la dynamique vésicale fonctionnelle indépendamment de la reconstruction génito-urinaire. Des explorations urodynamiques sont souvent réalisées en préopératoire pour déterminer la nécessité de gestes vésicaux concomitants tels qu’une appendicovésicostomie en cas de vidange incomplète et/ou d’une réimplantation urétérale en présence d’un RVU. Traditionnellement, trois interventions chirurgicales avec leurs modifications ont été rapportées dans la littérature pour corriger les défauts de la paroi abdominale associés au PBS.61,62,63 Alors que la procédure de Randolph consiste en l’excision d’une portion de la paroi abdominale inférieure afin de corriger la redondance fasciale verticale, les procédures de Monfort (Figure 4), (Figure 5), (Figure 6), (Figure 7), (Figure 8) et d’Ehrlich utilisent toutes deux un chevauchement vertical du fascia pour corriger la redondance latérale, tout en renforçant la paroi abdominale. Une approche laparoscopique de l’abdominoplastie a également été rapportée, s’avérant bénéfique à la fois pour aider à la reconstruction elle-même ainsi que pour délimiter les différents degrés de déficit musculaire.64 Il est également noté que les patients atteints de PBS peuvent chacun présenter des caractéristiques uniques quant à la variation de la sévérité et du schéma du déficit de la musculature abdominale, telle qu’une zone particulière présentant une absence complète de muscle. Smith et ses collègues ont récemment rapporté leurs adaptations de la procédure de Monfort, qui permettent une correction plus individualisée de la laxité de la paroi abdominale et le positionnement de l’ombilic à un emplacement anatomiquement plus correct.
Figure 4 Une incision médiane du processus xiphoïde au pubis, contournant l’ombilic.
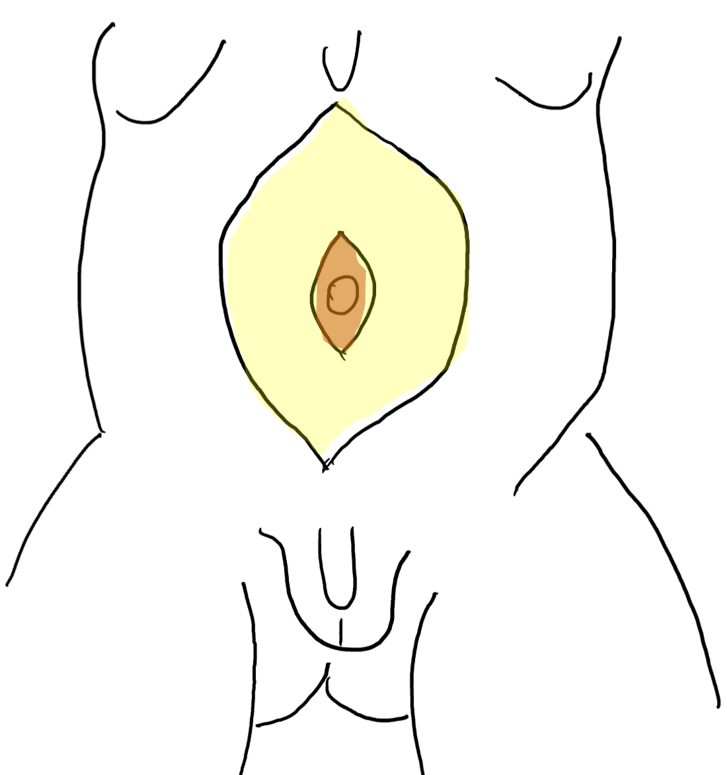
Figure 5 Décollement du lambeau cutané entre la graisse sous-cutanée et le plan aponévrotique. L’ombilic est soutenu par la bandelette aponévrotique centrale.
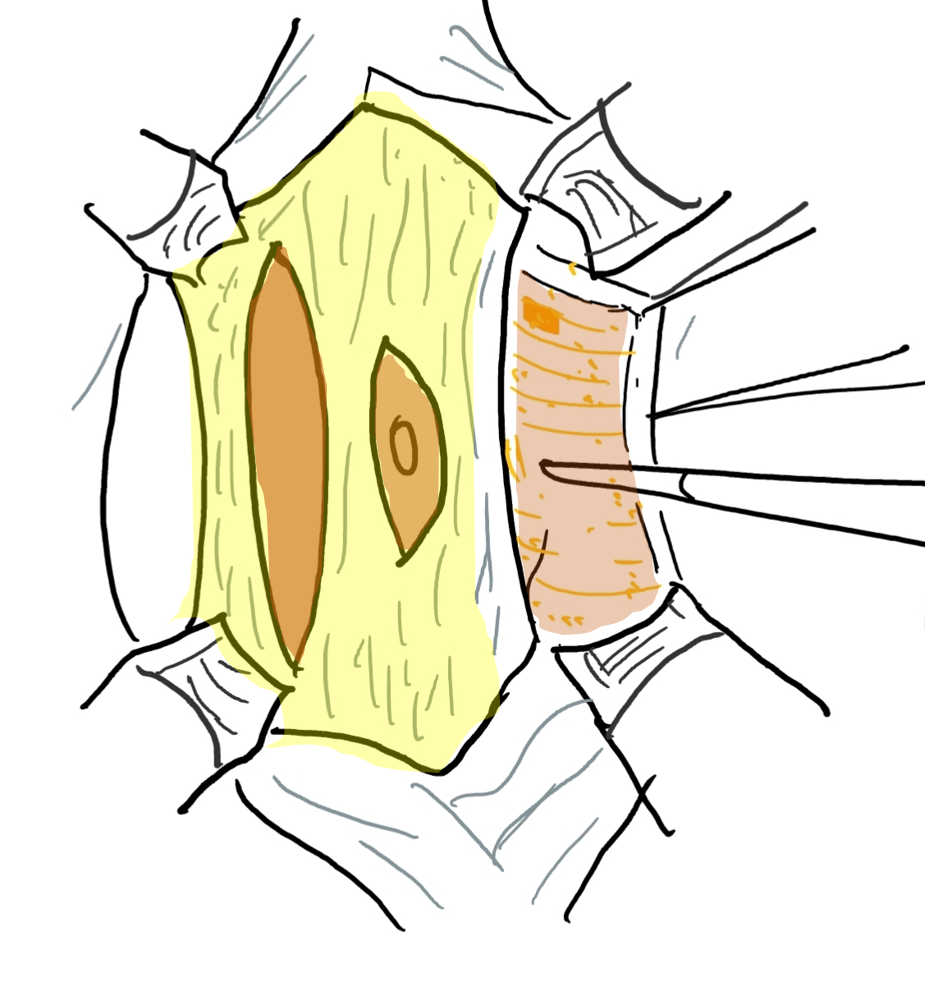
Figure 6 Le plan aponévrotique est ouvert par une incision latérale et parallèle au trajet des artères épigastriques supérieure et inférieure. L’extension latérale correspond à la ligne axillaire antérieure.
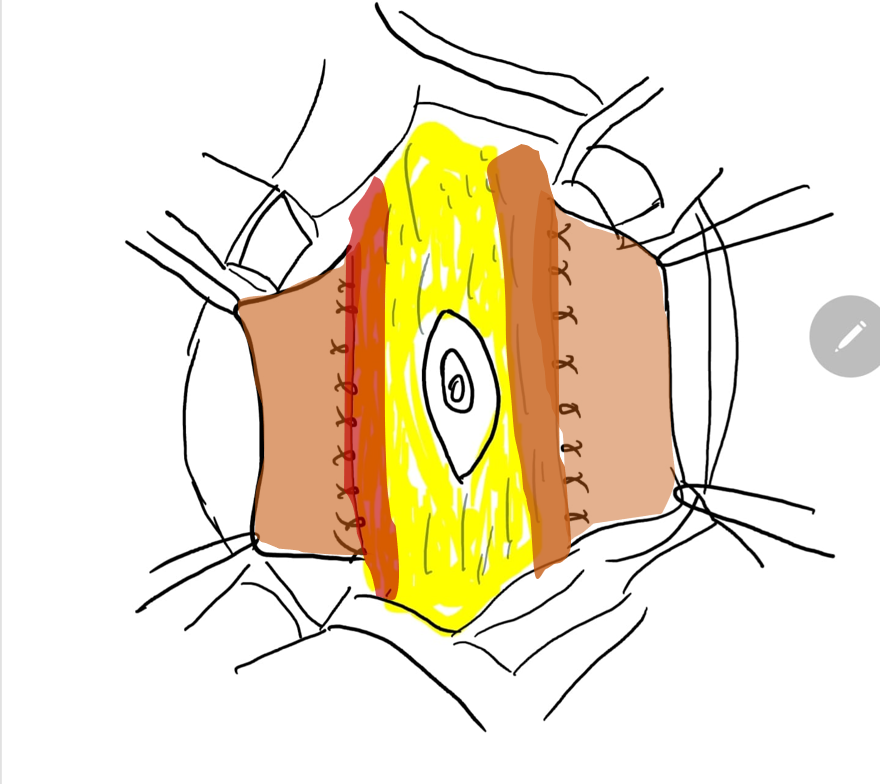
Figure 7 Le bord latéral de la bandelette aponévrotique centrale est suturé en profondeur sous le muscle.
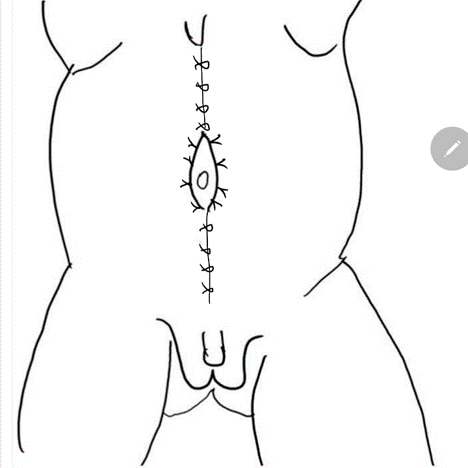
Figure 8 L’aponévrose latérale est ensuite fixée à la ligne médiane au-dessus et au-dessous de l’ombilic. La fermeture pantalon -sur- gilet assure un renfort supplémentaire et les lambeaux cutanés des deux côtés sont fermés sont fermés ensuite.
Cryptorchidie
Les testicules intra-abdominaux bilatéraux constituent une autre caractéristique emblématique du PBS. L’orchidopexie traditionnelle en un temps et l’orchidopexie de Fowler- Stephens en un temps ou en plusieurs temps sont autant d’options possibles pour la prise en charge des testicules, selon la longueur des vaisseaux gonadiques, et peuvent être réalisées par voie ouverte ou laparoscopique selon la préférence du chirurgien.65,66,67 La laxité de la paroi abdominale peut entraîner des difficultés techniques potentielles pour créer et maintenir un pneumopéritoine, qui peuvent être minimisées en utilisant des trocarts à expansion radiale et des débits de gaz élevés lors de l’orchidopexie laparoscopique, comme décrit par Philip et al..67,68 Il convient toutefois de souligner qu’en l’absence de tout problème cardiopulmonaire majeur, l’orchidopexie doit être réalisée le plus tôt possible comme le recommandent les actuelles [recommandations de l’AUA](https://www.auanet.org/guidelines/guidelines/cryptorchidism-guideline) et ne doit pas être retardée jusqu’à un âge plus avancé de l’enfance, afin d’augmenter les chances de fertilité et d’améliorer la probabilité d’une procédure en un seul temps, car la section des vaisseaux gonadiques est moins probable lorsque l’orchidopexie est réalisée précocement.
Reconstruction complète
La reconstruction complète chez le patient atteint du syndrome de prune-belly comprend une cystoplastie de réduction, la résection de l’uretère distal redondant et une réimplantation urétérale bilatérale après effilement.69 La reconstruction peut être associée à la réalisation d’une abdominoplastie et d’une orchidopexie bilatérale, généralement avant l’âge de deux ans. Cette approche agressive a été utilisée dans de nombreuses séries et a montré de bons résultats en termes de fonction rénale stable ou améliorée.
Dans l’abord décrit par Woodward, on réalise une large incision abdominale basse qui s’étend de l’extrémité de la 12e côte, le long de l’épine iliaque antéro-supérieure jusqu’au pubis, puis remonte en courbe jusqu’à la 12e côte du côté opposé. Cela tend à préserver l’innervation et la vascularisation segmentaires de la paroi abdominale conservée en vue de la reconstruction. De plus, lors de l’excision de la paroi abdominale redondante, on préserve la portion contenant le plus de fibres musculaires. L’exposition des voies urinaires obtenue avec cette incision est exceptionnelle. Cependant, l’utilisation de cette technique a diminué en raison de préoccupations concernant la section des vaisseaux épigastriques et le tissu abdominal latéral redondant. À la place, la plupart des chirurgiens privilégient l’abord de Monfort (Figure 4), (Figure 5), (Figure 6), (Figure 7), et (Figure 8)
Le péritoine postérieur est incisé le long du trajet des uretères. Les vaisseaux spermatiques sont mobilisés distalement; en veillant à ne pas compromettre les attaches péritonéales du canal déférent si une orchidopexie de Fowler-Stephens est nécessaire. La vessie est ouverte sur la ligne médiane au bistouri électrique. Les orifices urétéraux latéraux sont identifiés et chaque uretère est mobilisé. La grande longueur des uretères mobilisés permet la résection de leur tiers distal, habituellement la portion la plus anormale. On procède à un amincissement urétéral formel ou à une imbrication. Les uretères sont ensuite réimplantés, en utilisant une technique transtrigonale ou de Leadbetter Politano. La mise en place d’attelles urétérales sur les uretères opérés est réalisée, et celles-ci sont extériorisées à travers la vessie et par des incisions abdominales séparées.
Avant la cystoplastie de réduction, les testicules sont mobilisés avec un large tablier de péritoine postérieur. Dans certains cas, les testicules peuvent être adhérents à la vessie augmentée de volume et doivent être méticuleusement mobilisés afin de préserver la vascularisation. Si la reconstruction est réalisée avant l’âge de 2 ans, une orchidopexie standard avec des vaisseaux spermatiques intacts peut être pratiquée dans 80 % des cas. Sinon, une orchidopexie Fowler-Stephens différée ou formelle sera nécessaire
La taille de la vessie est réduite par résection du reliquat ourachal et du volumineux dôme vésical. Une bande d’épithélium vésical peut être retirée d’une des parois latérales et refermée selon la technique vest-over-pants. La vessie est drainée par une sonde de Malécot. Les testicules préalablement mobilisés sont amenés à travers la paroi abdominale distale au niveau du tubercule pubien. Une poche dartosienne est créée dans le scrotum et chaque testicule est fixé dans la poche par des sutures permanentes.
La paroi abdominale redondante est alors mise en tension et repérée sur la ligne médiane et latéralement aux angles costo-vertébraux afin de délimiter le tissu à exciser. Il est essentiel d’éviter l’ablation d’une quantité excessive de paroi abdominale. La seconde ligne d’incision est parallèle à l’incision précédente. Un large coin de paroi abdominale en pleine épaisseur est réséqué. La fermeture de la paroi abdominale débute par la mise en place de sutures clés avec des fils non résorbables de calibre 0 ou 1–0 au niveau des épines iliaques antérieures et de chaque tubercule pubien. Ces sutures doivent inclure le périoste de l’os et toute l’épaisseur du bord tissulaire proximal opposé. La tension ainsi créée permet parfois d’exciser en plus une redondance latérale de la paroi abdominale. Chaque segment entre les sutures clés est fermé par des points séparés affrontant toute l’épaisseur de la paroi abdominale. Le tissu sous-cutané est fermé pour prévenir les espaces morts et la peau est rapprochée par une suture intradermique résorbable.
Résultats à long terme
Qualité de vie
Bien que le pronostic global des patients atteints de PBS en termes de survie et de qualité de vie se soit considérablement amélioré, la PBS affecte profondément la qualité de vie liée à la santé des enfants concernés ainsi que celle de leurs aidants.70 Les patients présentent un fonctionnement physique, émotionnel, social et scolaire altéré et les aidants rapportent une qualité de vie globalement moindre en raison des fardeaux auxquels les familles d’enfants atteints de maladies chroniques sont souvent confrontées. Une autre étude a montré que les survivants atteints de PBS présentaient une incidence significativement élevée d’affections orthopédiques, gastro-intestinales et cardiopulmonaires qui altéraient leur qualité de vie, tandis que le traitement de ces affections non génito-urinaires améliore leur vie.70,71 Ces études soulignent que, en tant que responsable principal de la santé globale de ces patients médicalement complexes, l’urologue pédiatrique doit être conscient et préparé non seulement à traiter les complications génito-urinaires tardives de la PBS, mais aussi à évaluer et diagnostiquer les comorbidités non génito-urinaires qui influent directement sur leurs traitements médicaux et chirurgicaux ainsi que sur leur qualité de vie, lorsqu’ils passent de la puberté à l’âge adulte.
Insuffisance rénale et transplantation
Jusqu’à 30 % des patients, généralement ceux présentant d’emblée une altération de la fonction rénale, développent une insuffisance rénale chronique pendant l’enfance ou l’adolescence. L’insuffisance rénale terminale (IRT) précoce est considérée comme secondaire à une dysplasie rénale, tandis que l’insuffisance rénale survenant plus tard est souvent attribuée à des lésions parenchymateuses dues à des infections répétées et à l’augmentation de la pression transmise aux voies excrétrices supérieures, générée par une vidange incomplète.72 Yalcinkaya et al ont rapporté que l’âge médian au début de la suppléance rénale chez les garçons atteints de PBS était de 7 ans, ce qui était significativement plus jeune que chez les patients de sexe masculin atteints d’autres types de LUTO ou de dysplasie rénale, avec un âge médian à la première transplantation de seulement 9,3 ans.73
La transplantation rénale est nécessaire pour ces patients afin d’assurer une croissance et un développement normaux, et le succès de la transplantation chez les patients atteints de PBS peut être attendu comme équivalent à celui d’autres groupes appariés pour l’âge. Depuis le premier rapport en 1976, des reins de donneurs décédés et de donneurs vivants apparentés ont été transplantés avec succès chez des patients atteints de PBS, l’âge au moment de la transplantation allant de 8 mois à 21 ans.73,74 Avant la transplantation, des néphro-urétérectomies bilatérales sont habituellement réalisées dans tous les cas afin d’éviter des complications infectieuses. Une évaluation radiographique et urodynamique des voies urinaires basses est recommandée pour s’assurer de l’absence d’obstruction et d’une vidange vésicale équilibrée. Le recours au cathétérisme intermittent propre pour vider la vessie décompensée n’est pas une contre-indication à la transplantation rénale. Tous les patients doivent être maintenus sous antibioprophylaxie. Une complication inhabituelle de la transplantation dans le syndrome prune-belly est la torsion de l’allogreffe.75 La torsion, entraînant une perte du greffon, était due à l’absence de tonus de la paroi abdominale. La néphropexie a été recommandée pour éviter cette complication désastreuse. Dans une revue rétrospective de huit patients atteints du syndrome prune-belly ayant bénéficié d’une transplantation, il n’y avait pas de différence statistiquement significative concernant les décès des patients, la survie du greffon ou la fonction du greffon par rapport à des témoins appariés pour l’âge. Une analyse rétrospective plus récente d’une survie du greffon à 10 ans de 47% n’était pas statistiquement différente de celle des patients témoins appariés pour l’âge,73,74
Fonction vésicale
Comme mentionné précédemment, les études urodynamiques chez les patients atteints de PBS montrent généralement une compliance normale avec une sensibilité et une activité contractile diminuées. Par conséquent, certains patients ont tendance à retenir de grands volumes dans la vessie, dont la capacité augmente progressivement, avec un résidu post-mictionnel important. Le manque de soutien abdominal, l’éventuelle association d’une polyurie et l’attitude négative de certains patients à l’égard des mictions à heures fixes accentuent généralement cette tendance. Ainsi, pour prévenir une dilatation vésicale supplémentaire et une détérioration des voies urinaires supérieures, tous les patients, y compris ceux ayant bénéficié d’une reconstruction des voies urinaires basses et d’une abdominoplastie, doivent être entraînés le plus tôt possible et constamment encouragés à vider leur vessie à heures fixes, en y associant au besoin des mictions doubles ou triples et des manœuvres de Valsalva ou de Credé.76 En cas de déséquilibre progressif de la vidange vésicale, si un tel programme n’est pas réalisable ou s’avère inefficace, le cathétérisme vésical intermittent s’impose, soit par voie urétrale, soit via un canal de Mitrofanoff. À terme, une miction efficace est obtenue avec la croissance et l’amélioration de la sensation proprioceptive, ainsi qu’un meilleur soutien abdominal. Dans la série de Lopes et al, 17,4 % des patients ayant subi une reconstruction des voies urinaires ont nécessité un cathétérisme intermittent par l’urètre ou via un canal de Mitrofanoff. Rarement, certains de ces enfants retrouvent une miction normale avec une bonne vidange vésicale.
Croissance et développement musculosquelettique
Une croissance normale peut être attendue chez la plupart des patients ayant une fonction rénale normale, bien qu’un retard de croissance en l’absence d’atteinte rénale ait été observé chez un tiers des patients dans une série.34 Les déformations thoraciques, telles que le pectus excavatum, ne compromettent généralement pas la fonction pulmonaire chez l’enfant plus âgé et peuvent s’améliorer avec la croissance et l’exercice. Les patients atteints du syndrome peuvent présenter des anomalies musculosquelettiques altérant la fonction et l’esthétique, comme la scoliose, la lordose, ainsi que des anomalies de la cheville et de la hanche pouvant nécessiter une intervention orthopédique précoce. Dans une étude récente, les patients plus âgés et leurs familles ont été informés que la correction des problèmes musculosquelettiques par abdominoplastie et/ou interventions orthopédiques était le moyen le plus courant pour les professionnels de santé d’améliorer la qualité de vie de leurs patients.
Fonction sexuelle et fertilité
Un schéma normal du développement sexuel secondaire peut être attendu, la production hormonale par les testicules étant préservée, mais la fonction sexuelle peut être altérée par une éjaculation rétrograde. L’infertilité primaire est généralement la règle et, lorsqu’elle survient, elle serait due à une combinaison d’anomalies histologiques testiculaires avec altération de la spermatogenèse, de défauts structurels des conduits, et d’anomalies prostatiques entraînant une éjaculation rétrograde.77 Cependant, la paternité est possible grâce aux techniques de procréation assistée (ART), en particulier chez ceux ayant bénéficié d’une orchidopexie précoce réussie, et des naissances vivantes normales ont été rapportées chez des patients adultes atteints de PBS classique, rendues possibles par des techniques de prélèvement des spermatozoïdes et l’injection intracytoplasmique de spermatozoïdes.78,79,80 Une grossesse normale avec accouchement vaginal assisté a également été décrite chez une patiente atteinte du syndrome.81
Conclusions
Comme pour la plupart des malformations congénitales complexes, la clé de la prise en charge du PBS est une approche pluridisciplinaire, basée sur le travail d’équipe, offrant des soins individualisés. Compte tenu du large spectre de la maladie, le traitement du syndrome de Prune Belly est spécifique à chaque patient. Lorsque l’infection et la stase compromettent la croissance ou la fonction rénales, une intervention chirurgicale agressive visant à favoriser le drainage est recommandée. Chez ces nourrissons, les testicules sont placés précocement dans le scrotum afin d’améliorer la fertilité, ce qui peut être réalisé par des techniques laparoscopiques ou ouvertes. En raison de l’évolution des conditions urodynamiques des voies urinaires inférieures et de leurs effets sur la fonction rénale et l’infection urinaire, tous les patients atteints du syndrome de Prune Belly nécessitent une surveillance urologique attentive tout au long de la vie.
Points clés
- Le syndrome de prune-belly (PBS) inclut une constellation d’anomalies de sévérité variable. Les trois principales constatations sont un déficit de la musculature abdominale, des testicules intra-abdominaux bilatéraux et des anomalies des voies urinaires.
- Les voies urinaires sont caractérisées par des degrés variables d’hydronéphrose, de dysplasie rénale, des uretères dilatés et tortueux, une vessie augmentée de volume, et un urètre prostatique dilaté/ mégalourètre.
- D’autres anomalies associées impliquent l’appareil respiratoire, le tractus gastro-intestinal, le système cardiaque et l’appareil musculo-squelettique.
- Le déterminant le plus important de la survie à long terme est généralement la sévérité de l’anomalie des voies urinaires, en particulier le degré de dysplasie rénale.
- L’hydronéphrose non obstructive est la règle. C’est l’infection rénale plutôt que l’obstruction qui représente le plus grand risque pour la fonction rénale.
- L’intervention urologique initiale est dirigée vers le drainage vésical et une vidange vésicale adéquate afin d’éviter les infections des voies urinaires (IVU) et ainsi préserver la fonction rénale.
- La prise en charge chirurgicale prend la forme d’une reconstruction des voies urinaires, d’une abdominoplastie et d’une orchidopexie bilatérale, qui peuvent être réalisées séparément ou en un seul temps chez les enfants plus jeunes.
- La prise en charge des patients atteints de PBS nécessite une large équipe multidisciplinaire pour aider ces enfants à s’épanouir, à prendre du poids et à être préparés à une chirurgie urologique si nécessaire.
Références
- Woodard S JR, E.A.. Prune belly syndrome.
- Denes FT, Lopes RI. Prune-belly syndrome. In: Partin AW, Dmochowski RR, Kavoussi LR, Peters CA, editors. Campbell wals-wein urology. 12th ed. Philadelphia: Elsevier, Inc; 2021. DOI: 10.25060/residpediatr-2018.v8n1-07.
- Grimsby GM, Harrison SM, Granberg CF, Bernstein IH, Baker LA. Impact and frequency of extra-genitourinary manifestations of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 280 281–286. DOI: 10.1016/j.jpurol.2015.06.005.
- Wong DG, Arevalo MK, Passoni NM, Iqbal NS, Jascur T, Kern AJ. Phenotypic severity scoring system and categorization for prune belly syndrome: application to a pilot cohort of 50 living patients. BJU Int 2019; 123 (1): 130–139. DOI: 10.1111/bju.14524.
- Wheatley JM, Stephens FD, Hutson JM. Prune-belly syndrome: ongoing controversies regarding pathogenesis and management. Semin Pediatr Surg 1996; 5 (2): 95–106.
- Gonzalez R, Reinberg Y, Burke B, Wells T, Vernier RL. Early bladder outlet obstruction in fetal lambs induces rental dysplasia and the prune-belly syndrome. J Pediatr Surg 1990; 25 (3): 342–345. DOI: 10.1016/0022-3468(90)90083-l.
- Ramasamy R, Haviland M, Woodard B JR, J.G.. Patterns of inheritance in familial prune belly syndrome. Urology 2005; 65 (6). DOI: 10.1016/j.urology.2004.12.050.
- Reinberg Y, Shapiro E, Manivel JC, Manley CB, Pettinato G, Gonzalez R. Prune belly syndrome in females: A triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr 1991; 118 (3). DOI: 10.1016/s0022-3476(05)82153-5.
- Balaji KC, Patil A, Townes PL, Primack W, Skare J, Hopkins T. Concordant prune belly syndrome in monozygotic twins. Urology 2000; 55 (6). DOI: 10.1016/s0090-4295(00)00452-0.
- Iqbal NS, Jascur TA, Harrison SM, Edwards AB, Smith LT, Choi ES. Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene. BMC Med Genet 2020; 21 (1). DOI: 10.1186/s12881-020-0973-x.
- Granberg CF, Harrison SM, D D. Genetic basis of prune belly syndrome: Screening for HNF1beta gene. J Urol 2012; 187 (1).
- Stephens FD, Gupta D. Pathogenesis of the prune belly syndrome. J Urol 1994; 152: 2328–2331. DOI: 10.1016/s0022-5347(17)31669-5.
- Palmer JM, Tesluk H. Ureteral pathology in the prune belly syndrome. J Urol 1974; 111 (5). DOI: 10.1016/s0022-5347(17)60050-8.
- Gearhart JP, Lee BR, Partin AW, Epstein JI, Gosling JA, Kogan BA. A quantitative histological evaluation of the dilated ureter of childhood. II: Ectopia, posterior urethral valves and the prune belly syndrome. J Urol 1995; 153 (1). DOI: 10.1097/00005392-199501000-00069.
- Snyder HM, Harrison NW, Whitfield HN, Williams I. Urodynamics in the prune belly syndrome. Br J Urol 1976; 48 (7): 663–670. DOI: 10.1111/j.1464-410x.1976.tb06716.x.
- Kinahan TJ, Churchill BM, McLorie GA, Gilmour RF, Khoury AE. The efficiency of bladder emptying in the prune belly syndrome. J Urol 1992; 148 (2 Pt 2): 600–603. DOI: 10.1016/s0022-5347(17)36665-x.
- Favorito LA, Pires RS, Gallo CM, Sampaio FJB. Study of prostate growth in prune belly syndrome and anencephalic fetuses. J Pediatr Surg 2020; 55 (10): 2221–2225. DOI: 10.1016/j.jpedsurg.2019.10.054.
- Volmar KE, Fritsch MK, Perlman EJ, Hutchins GM. Patterns of congenital lower urinary tract obstructive uropathy: Relation to abnormal prostate and bladder development and the prune belly syndrome. Pediatr Dev Pathol 2001; 4 (5). DOI: 10.1007/s10024001-0042-1.
- Deklerk DP, Scott WW. Prostatic Maldevelopment in the prune belly syndrome: A defect in prostatic stromal-epithelial interaction. J Urol 1978; 120 (3). DOI: 10.1016/s0022-5347(17)57168-2.
- Cabral B, Majidi A, Gonzalez R. Ectopic vasa deferentia in an infant with the prune belly syndrome. J Urol 1988; 94 (4).
- Kroovand RL, Al-Ansari RM, Perlmutter AD. Urethral and genital malformations in prune belly syndrome. J Urol 1982; 127 (1): 94–96. DOI: 10.1016/s0022-3468(82)80571-x.
- Berdon WE, Baker DH, Wigger HJ, Blanc WA. The radiologic and pathologic spectrum of the prune belly syndrome. The importance of urethral obstruction in prognosis. Radiol Clin North Am 1977; 15 (1).
- Hoagland MH, Hutchins GM. Obstructive lesions of the lower urinary tract in the prune belly syndrome. J Urol 1987; 138 (2). DOI: 10.1016/s0022-5347(17)43220-4.
- Volmar KE, Nguyen TC, Holcroft CJ, Blakemore KJ, Hutchins GM. Phimosis as a cause of the prune belly syndrome: Comparison to a more common pattern of proximal penile urethra obstruction. Virchows Arch 2003; 442 (2). DOI: 10.1007/s00428-002-0730-x.
- Beasley S, Bettenay F, Hutson J. The anterior urethra provides clues to the aetiology of prune belly syndrome. Pediatr Surg Int 1988; 3–3(2–3. DOI: 10.1007/bf00182775.
- Sellers BB, McNeal R, Smith RV, Griswold WR, Mendoza SA. Congenital megalourethra associated with prune belly syndrome. J Urol 1976; 116 (6). DOI: 10.1016/s0022-5347(17)59027-8.
- Uehling DT, Zadina SP, Gilbert E. Testicular histology in triad syndrome. Urology 1984; 23 (4). DOI: 10.1016/0090-4295(84)90141-9.
- Massad CA, Cohen MB, Kogan BA, Beckstead JH. Morphology and histochemistry of infant testes in the prune belly syndrome. J Urol 1991; 146 (6): 1598–1600. DOI: 10.1016/s0022-5347(17)38178-8.
- Orvis BR, Bottles K, Kogan BA. Testicular histology in fetuses with the prune belly syndrome and posterior urethral valves. J Urol 1988; 139 (2): 335–337. DOI: 10.1016/s0022-5347(17)42403-7.
- Sayre R, Stephens R, Chonko AM. Prune belly syndrome and retroperitoneal germ cell tumor. Am J Med 1986; 81 (5). DOI: 10.1016/s0022-5347(17)44221-2.
- Woodhouse CR, Ransley PG. Teratoma of the testis in the prune belly syndrome. Br J Urol 1983; 55 (5).
- Mininberg DT, Montoya F, Okada K, Galioto F, Presutti R. Subcellular muscle studies in the prune belly syndrome. J Urol 1973; 109 (3). DOI: 10.1097/00006534-197311000-00052.
- Afifi AK, Rebeiz J, Mire J, Andonian SJ, Kaloustian VM. The myopathology of the prune belly syndrome. J Neurol Sci 1972; 15 (2). DOI: 10.1016/0022-510x(72)90003-2.
- Loder RT, Guiboux JP, Bloom DA, Hensinger RN. Musculoskeletal aspects of prune-belly syndrome. Description and Pathogenesis Am J Dis Child 1992; 146 (10): 1224–1229. DOI: 10.1001/archpedi.1992.02160220110034.
- Geary DF, MacLusky IB, Churchill BM, McLorie G. A broader spectrum of abnormalities in the prune belly syndrome. J Urol 1986; 135 (2). DOI: 10.1016/s0022-5347(17)45627-8.
- Alford BA, Peoples WM, Resnick JS, L’Heureux PR. Pulmonary complications associated with the prune-belly syndrome. Radiology 1978; 129 (2). DOI: 10.1148/129.2.401.
- Wright B JR, RF N, JC P, ET S, ME S, L.E.. Gastrointestinal malformations associated with prune belly syndrome: Three cases and a review of the literature. Pediatr Pathol 1986; 5 (3–4). DOI: 10.3109/15513818609068868.
- Salihu HM, Tchuinguem G, Aliyu MH, Kouam L. Prune belly syndrome and associated malformations. A 13-year experience from a developing country. West Indian Med J 2003; 52 (4).
- Brinker MR, Palutsis RS, Sarwark JF. The orthopaedic manifestations of prune-belly (Eagle-Barrett) syndrome. J Bone Joint Surg 1995; 77 (2). DOI: 10.2106/00004623-199502000-00012.
- Cazorla E, Ruiz F, Abad A, Monleon J. Prune belly syndrome: Early antenatal diagnosis. Eur J Obstet Gynecol Reprod Biol 1997; 72 (1). DOI: 10.1016/s0301-2115(96)02664-4.
- Fitzsimons RB, Keohane C, Galvin J. Prune belly syndrome with ultrasound demonstration of reduction of megacystis in utero. Br J Radiol 1985; 58 (688). DOI: 10.1259/0007-1285-58-688-374.
- Aaronson IA. Posterior urethral valve masquerading as the prune belly syndrome. Br J Urol 1983; 55 (5). DOI: 10.1016/s0022-3468(84)80160-8.
- Tonni G, Ida V, Alessandro V, Bonasoni MP. Prune-belly syndrome: case series and review of the literature regarding early prenatal diagnosis, epidemiology, genetic factors, treatment, and prognosis. Fetal Pediatr Pathol 2013; 31 (1): 13–24. DOI: 10.3109/15513815.2012.659411.
- Yamamoto H, Nishikawa S, Hayashi T, Sagae S, Kudo R. Antenatal diagnosis of prune belly syndrome at 11 weeks of gestation. J Obstet Gynaecol Res 2001; 27 (1). DOI: 10.1111/j.1447-0756.2001.tb01213.x.
- Seidel NE, Arlen AM, Smith EA, Kirsch AJ. Clinical manifestations and management of prune-belly syndrome in a large contemporary pediatric population. Urology 2015; 85 (1). DOI: 10.1016/j.urology.2014.09.029.
- Woods AG, Brandon DH. Prune belly syndrome. A focused physical assessment. Adv Neonatal Care 2007; 7 (3): 144–145.
- Arlen AM, Nawaf C, Kirsch AJ. Prune belly syndrome: current perspectives. Pediatric Health Med Ther 2019; 10: 75–81. DOI: 10.2147/phmt.s188014.
- Rushton HG, Majd M, Chandra R, Yim D. Evaluation of 99M technetium-dimercapto-succinic acid renal scans in experimental acute pyelonephritis in piglets. J Urol 1988; 140 (5). DOI: 10.1016/s0022-5347(17)41992-6.
- Garcia-Roig ML, Grattan-Smith JD, Arlen AM, Smith EA, Kirsch AJ. Detailed evaluation of the upper urinary tract in patients with prune belly syndrome using magnetic resonance urography. J Pediatr Urol 2016; 12 (2). DOI: 10.1016/j.jpurol.2015.11.008.
- Kaplan BS. In utero intervention in prune-belly syndrome. Pediatr Nephrol 1999; 13 (2). DOI: 10.1007/s004670050581.
- Makino Y, Kobayashi H, Kyono K, Oshima K, Kawarabayashi T. Clinical results of fetal obstructive uropathy treated by vesicoamniotic shunting. Urology 2000; 55 (1). DOI: 10.1016/s0090-4295(99)00403-3.
- Burbige KA, Amodio J, Berdon WE, Hensle TW, Blanc W, Lattimer JK. Prune belly syndrome: 35 years of experience. J Urol 1987; 137 (1). DOI: 10.1016/s0022-5347(17)43880-8.
- Fallat ME, Skoog SJ, Belman AB, Eng G, Randolph JG. The prune belly syndrome: A comprehensive approach to management. J Urol 1989; 142 (3). DOI: 10.1016/s0022-5347(17)38895-x.
- Tank ES, McCoy G. Limited surgical intervention in the prune belly syndrome. J Pediatr Surg 1983; 18 (6). DOI: 10.1016/s0022-3468(83)80004-9.
- Woodhouse CRJ, Kellett MJ, Williams DI. Minimal surgical interference in the prune belly syndrome. Br J Urol 1979; 51 (6). DOI: 10.1111/j.1464-410x.1979.tb03582.x.
- Woodard P JR, T.S.. Reconstruction of the urinary tract in prune belly uropathy. J Urol 1978; 119 (6). DOI: 10.1016/s0022-5347(17)57644-2.
- Bukowski TP, Perlmutter AD. Reduction cystoplasty in the prune belly syndrome: a long-term followup. J Urol 1994; 152 (6 Pt 1): 2113–2116. DOI: 10.1016/s0022-5347(17)32333-9.
- Wille MA, Jayram G, Gundeti MS. Feasibility and early outcomes of robotic-assisted laparoscopic Mitrofanoff appendicovesicostomy in patients with prune belly syndrome. BJU Int 2011; 109 (1). DOI: 10.1111/j.1464-410x.2011.10317.x.
- Ehrlich RM, Lesavoy MA, Fine RN. Total abdominal wall reconstruction in the prune belly syndrome. J Urol 1986; 136: 282–285. DOI: 10.1016/s0022-5347(17)44842-7.
- Randolph J, Cavett C, Eng G. Abdominal wall reconstruction in the prune belly syndrome. J Pediatr Surg 1981; 16 (6): 960–964. DOI: 10.1016/s0022-5347(17)52848-7.
- Monfort G, Guys JM, Bocciardi A, Coquet M, Chevallier D. A novel technique for reconstruction of the abdominal wall in the prune belly syndrome. J Urol 1991; 146 (2 Pt 2): 639–640. DOI: 10.1016/s0022-5347(17)37880-1.
- Denes FT, Lopes RI, Oliveira LM, Tavares A, Srougi M. Modified abdominoplasty for patients with the Prune Belly syndrome. Urology 2014; 83 (2): 451–454. DOI: 10.1016/j.urology.2013.09.031.
- Fearon JA, Varkarakis G. Dynamic abdominoplasty for the treatment of prune belly syndrome. Plast Reconstr Surg 2012; 130 (3). DOI: 10.1097/prs.0b013e31825dc170.
- Franco I. Laparoscopic assisted modification of the firlit abdominal wall plication. J Urol 2005; 174 (1). DOI: 10.1097/01.ju.0000161214.65458.c2.
- Patil KK, Duffy PG, Woodhouse CR, Ransley PG. Long-term outcome of Fowler-Stephens orchiopexy in boys with prune belly syndrome. J Urol 2004; 171 (4): 1666–1669. DOI: 10.1097/01.ju.0000118139.28229.f5.
- Yu T-J, Lai M-K, Chen W-F, Wan Y-L. Two-stage orchiopexy with laparoscopic clip ligation of the spermatic vessels in prune-belly syndrome. J Pediatr Surg 1995; 30 (6). DOI: 10.1016/0022-3468(95)90768-8.
- Philip J, Mullassery D, Craigie RJ, Manikandan R, Kenny SE. Laparoscopic orchidopexy in boys with prune belly syndrome—Outcome and technical considerations. J Endourol 2011; 25 (7). DOI: 10.1089/end.2010.0257.
- Saxena AK, Brinkmann OA. Unique features of prune belly syndrome in laparoscopic surgery. J Am Coll Surg 2007; 205 (2). DOI: 10.1016/j.jamcollsurg.2007.03.008.
- Lopes RI, Tavares A, Srougi M, Dénes FT. 27 years of experience with the comprehensive surgical treatment of prune belly syndrome. J Pediatr Urol 2015; 11 (5): 276 271–277. DOI: 10.1016/j.jpurol.2015.05.018.
- Arlen AM, Kirsch SS, Seidel NE, Garcia-Roig M, Smith EA, Kirsch AJ. Health-related quality of life in children with Prune belly Syndrome and their caregivers. Urology 2016; 87 (224). DOI: 10.1016/j.urology.2015.09.028.
- Lesavoy MA, Chang EI, Suliman A, Taylor J, Kim SE, Ehrlich RM. Long-term follow-up of total abdominal wall reconstruction for prune belly syndrome. Plast Reconstr Surg 2012; 129 (1). DOI: 10.1097/prs.0b013e3182362091.
- Reinberg Y, Manivel JC, Pettinato G, Gonzalez R. Development of renal failure in children with the prune belly syndrome. J Urol 1991; 145 (5): 1017–1019. DOI: 10.1016/0022-3468(92)90159-5.
- Alcinkaya F, Bonthuis M, Erdogan BD, Stralen KJ, Baiko S, Chehade H, et al.. Outcomes of renal replacement therapy in boys with prune belly syndrome: findings from the ESPN/ERA-EDTA Registry. Pediatr Nephrol 2018; 33 (1): 117–124. DOI: 10.1007/s00467-017-3770-9.
- Fontaine E, Salomon L, Gagnadoux MF, Niaudet P, Broyer M, Beurton D. Long-term results of renal transplantation in children with the prune-belly syndrome. J 1997; Urol.158(3 Pt 1):892-4. DOI: 10.1097/00005392-199709000-00067.
- Marvin RG, Halff GA, Elshihabi I. Renal allograft torsion associated with prune-belly syndrome. Pediatr Nephrol 1995; 9 (1). DOI: 10.1007/bf00858981.
- Smith CA, Smith EA, Parrott TS, Broecker BH, Woodard JR. Voiding function in patients with the prune-belly syndrome after Monfort abdominoplasty. J Urol 1998; 159 (5). DOI: 10.1016/s0022-3468(99)90298-1.
- Woodhouse CR, Snyder HM. Testicular and sexual function in adults with prune belly syndrome. J Urol 1985; 133 (4): 607 9. DOI: 10.1016/s0022-5347(17)49108-7.
- Halpern JA, Das A, Brannigan RE. Successful sperm retrieval in prune belly syndrome. Asian J Urol 2020; 7 (4): 376–378. DOI: 10.1016/j.ajur.2019.07.004.
- Kolettis PN, Ross JH, Kay R, Thomas AJ. Sperm retrieval and intracytoplasmic sperm injection in patients with prune-belly syndrome. Fertil Steril 1999; 72 (5). DOI: 10.1016/s0015-0282(99)00388-x.
- Fleming SD, Varughese E, Hua VK, Robertson A, Dalzell F, Boothroyd CV. Normal live births after intracytoplasmic sperm injection in a man with the rare condition of Eagle-Barrett syndrome (prune-belly syndrome. Fertil Steril 2013; 100 (6). DOI: 10.1016/j.fertnstert.2013.07.1994.
- Hillman RT, Garabedian MJ, Wallerstein RJ. Pregnancy outcome in a woman with prune belly syndrome. BMJ Case Reports 2012. DOI: 10.1136/bcr-2012-006490.
Dernière mise à jour: 2025-09-22 07:59