
23: Mises à jour dans la prise en charge de l'exstrophie
Ce chapitre prendra environ 20 minutes de lecture.
Introduction
Le complexe exstrophie-épispadias (CEE) comprend un large spectre d’anomalies congénitales, toutes issues du même défaut embryologique, allant de l’épispadias glandulaire simple à l’exstrophie cloacale.1
Épidémiologie
Le complexe exstrophie vésicale-épispadias-exstrophie cloaquale (BEEC) est plus fréquent chez les sujets caucasiens. L’exstrophie vésicale classique (BEX) est le sous-type prédominant (50 %), survenant dans 2,15 - 3,3/100 000 naissances vivantes, et l’exstrophie cloaquale (CEX) est observée dans 1/200 000. Au cours des dernières décennies, on observe une augmentation de la suspicion diagnostique avec les progrès de la médecine fœtale.2,3
On observe une prédominance masculine et le rapport hommes:femmes a été rapporté entre 2,3-6:1.4 Cependant, l’exstrophie cloaquale est identique dans les deux sexes (1:1).
La mortalité chez les patients atteints d’exstrophie vésicale est faible (4 %). Grâce aux progrès des techniques chirurgicales et à la prise en charge actuelle, la survie à long terme de ces patients est excellente, avec des améliorations de la fertilité et de la fonction sexuelle tant chez les femmes que chez les hommes atteints de BEX. En outre, la survie des enfants atteints de CEX s’est améliorée, passant de 50 % en 1960 à plus de 80 % aujourd’hui.
Étiopathogénie
Embryologie
La séparation du cloaque primitif en sinus urogénital et intestin postérieur se produit au cours du premier trimestre de la grossesse, à peu près au moment où se forme la paroi abdominale antérieure. La membrane cloacale est une couche bilaminaire située à l’extrémité caudale du disque germinatif qui occupe la paroi abdominale infra-ombilicale. La pénétration mésodermique entre les couches ectodermique et endodermique de la membrane cloacale bilaminaire entraîne la formation de la musculature abdominale inférieure et des os pelviens. Après la pénétration mésenchymateuse, le septum urorectal progresse caudalement et divise le cloaque en vessie en avant et rectum en arrière. Distalement, le septum rejoint le reliquat postérieur de la membrane bilaminaire, qui finit par se perforer et former les orifices urogénital et anal. Les tubercules génitaux appariés migrent médialement et fusionnent sur la ligne médiane, en position céphalique par rapport à la membrane dorsale avant la perforation. Un défaut de migration des cellules mésenchymateuses entre les couches ectodermique et endodermique de la paroi abdominale inférieure entraîne une instabilité de la membrane cloacale.5 La rupture prématurée de cette membrane, avant sa migration caudale, conduit au développement de ce groupe d’anomalies infra-ombilicales. L’étendue du défaut infra-ombilical et le stade de développement au moment de la rupture déterminent si l’issue est une extrophie vésicale, une extrophie cloacale ou un épispadias. Si la rupture survient après la séparation complète des voies génito-urinaires et du tractus gastro-intestinal, une BEX classique se produit. Cependant, si cela survient avant la descente du septum urorectal, il y a externalisation des voies urinaires inférieures et de la portion distale du tractus gastro-intestinal, donnant naissance à une extrophie cloacale (Figure 1).
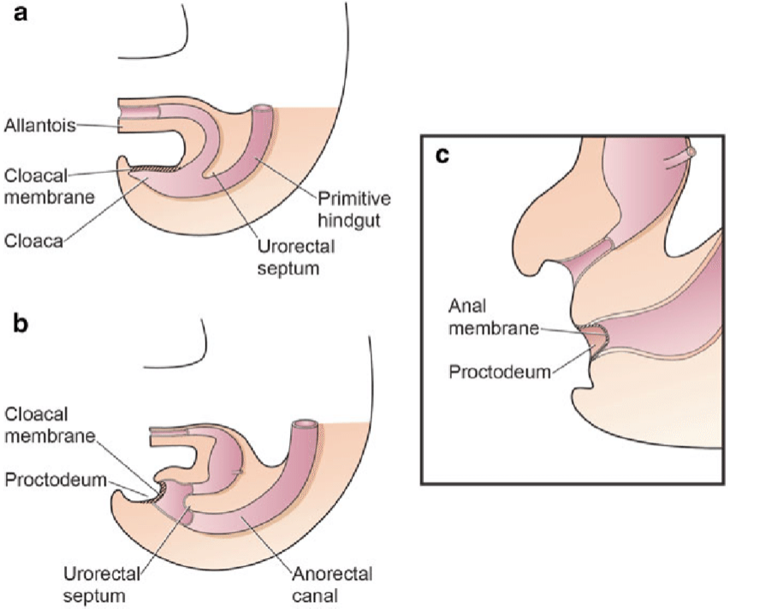
Figure 1 Division du cloaque en un sinus urogénital primitif antérieur et un rectum postérieur entre la 4e et la 6e semaine de gestation
La théorie la plus couramment admise du développement embryonnaire de l’exstrophie, soutenue par Marshall et Muecke,6 décrit le défaut fondamental comme un surdéveloppement anormal de la partie inférieure de la membrane cloacale, qui empêche la migration médiale du tissu mésenchymateux. Par conséquent, le développement adéquat de la paroi abdominale ne se produit pas. Le moment de la rupture de ce défaut cloacal détermine la gravité de l’affection. Les perforations centrales entraînant une exstrophie classique ont l’incidence la plus élevée (60%) tandis que les variantes d’exstrophie représentent 30% et l’exstrophie cloacale 10%.
D’autres théories sont avancées concernant la cause du complexe exstrophie-épispadias.7,8 Ambrose et O’Brian9 ont postulé qu’un développement anormal des bourgeons génitaux, avec une fusion sur la ligne médiane en dessous plutôt qu’au-dessus de la membrane cloaquale, entraîne le défaut d’exstrophie. Une autre hypothèse décrit une insertion caudale anormale du pédicule embryonnaire avec défaut d’interposition du tissu mésenchymateux sur la ligne médiane.10 En raison de ce défaut, la translocation du cloaque dans les profondeurs de la cavité abdominale ne se produit pas. Une membrane cloaquale qui demeure dans une position infra-ombilicale superficielle représente un état embryonnaire instable avec une forte tendance à se désintégrer.11 Aucune théorie ne semble élucider tous les aspects du complexe observé cliniquement et des études supplémentaires sont en cours pour décrire pleinement le processus de développement qui aboutit en fin de compte à la formation du complexe exstrophie-épispadias.
Étiologie
L’étiologie exacte de cette maladie n’a pas encore été identifiée. Néanmoins, il a été confirmé que l’événement déclencheur survient tôt au cours de la grossesse.8 Plusieurs études en cours suggèrent une possible base génétique pour le développement de ces affections.11 On observe une incidence plus élevée chez les enfants de mères ayant reçu de fortes doses de progestérone aux premiers stades de la grossesse, comme dans les cas de traitements de procréation assistée, et il est estimé que l’incidence est jusqu’à 7,5 fois plus élevée dans les cas où la fécondation in vitro a été utilisée.11
Risque de récidive
Le rôle possible de facteurs génétiques impliqués dans l’expression de l’EEC repose sur l’augmentation du risque de récurrence chez la descendance des individus atteints.8 Parmi les frères et sœurs, Ives et al ont estimé le risque de récurrence à environ 1 % chez des parents non consanguins et non atteints de cas de BEX.12 Shapiro et al.13 ont estimé le risque de récurrence pour les frères et sœurs d’origine européenne pour une BEX isolée à 1 sur 275, après avoir enquêté 2,500 familles CEB. Ils ont également décrit un risque 400 fois plus élevé de BEX classique chez la descendance d’individus atteints par rapport à la population générale. En se basant sur une prévalence de 3.3:100,000 (~1:30.000) pour la BEX isolée parmi les populations d’origine européenne, le rapport du risque de récurrence chez les frères et sœurs a été calculé à 108 (1:275/1:30.000 ~108).14 Bien que les patientes atteintes d’EEC représentent la minorité des patients, fait intéressant, seules les femmes atteintes ont eu une descendance atteinte.13 Parmi d’autres explications, ce risque de récurrence apparemment plus élevé chez les femmes atteintes pourrait être dû à une plus grande susceptibilité génétique à l’EEC (appelé effet de Carter).15
Récurrences familiales
Bien que la survenue familiale soit rare, 30 familles multiplex rapportées étayent l’idée d’une susceptibilité génétique sous-jacente à l’EEC.16 Dans la plupart de ces familles, deux membres sont atteints. Cependant, dans de rares cas, la transmission de l’EEC peut être compatible avec une hérédité autosomique dominante à pénétrance réduite ou avec un caractère autosomique récessif ou une transmission liée à l’X.17 Ces observations indiquent qu’un ou plusieurs gènes ayant un effet majeur sur le phénotype existent, bien que dans la plupart des cas des facteurs causaux supplémentaires soient nécessaires pour que le phénotype se manifeste. Un petit sous-groupe de cas peut suivre une hérédité mendélienne tandis que dans la majorité, l’EEC est héritée comme un caractère complexe avec de multiples facteurs génétiques (héréditaires ou *de novo *mutations somatiques ou de la lignée germinale), et des interactions complexes gène-gène ou gène-environnement contribuant à sa formation.8
Génétique moléculaire de l’EEC
Des analyses cytogénétiques et moléculaires ont révélé des anomalies chromosomiques chez 20 patients atteints d’EEC à ce jour, bien qu’aucune ne semble être causale.8 Des aberrations chromosomiques numériques ont été observées chez six patients. Par ailleurs, chez quatre patients de sexe masculin atteints de BEX, une patiente atteinte de BEX et une fille atteinte de CEX, une association avec la trisomie 21 a été retrouvée.16 L’aneuploïdie des chromosomes sexuels dans cinq de ces cas pourrait indiquer l’existence d’un locus gonosomique impliqué dans la formation de l’EEC. Des aberrations structurelles ont été identifiées dans six cas d’EEC et chez un patient présentant simultanément une CEX et une hypomélanose d’Ito. Bien que les points de cassure exacts n’aient été déterminés dans aucun de ces cas, plusieurs translocations impliquant la région q32-ter du chromosome 9 ont été détectées.
Agents tératogènes et EEC
Les études sur les jumeaux et les données épidémiologiques suggèrent que des facteurs environnementaux jouent un rôle dans l’étiologie de l’EEC. Cependant, les études épidémiologiques existantes n’ont pas identifié de facteurs tératogènes majeurs.4,12,18 Plusieurs études ont confirmé que le sexe masculin, la race, l’âge parental avancé4 et l’augmentation de la parité, même après ajustement pour l’âge,19 sont des facteurs de risque prédisposants. Gambhir et al.18 ont décrit que l’exposition maternelle périconceptionnelle au tabagisme était significativement plus fréquente chez les patients atteints de CEX que dans un groupe combiné de patients présentant un épispadias et une BEX classique. Plusieurs rapports ont décrit la survenue d’EEC chez des nourrissons issus d’une *in vitro *fécondation, mais il reste débattu de savoir si l’incidence des enfants atteints d’EEC conçus par *in vitro *fécondation est plus élevée que prévu.8,20
Présentation clinique
Période anténatale
Malgré l’importance de l’anomalie de la paroi abdominale inférieure et du développement des organes pelviens, l’exstrophie vésicale reste difficile à diagnostiquer de manière fiable à l’échographie prénatale.21 Cela est probablement dû à sa faible incidence et au fait qu’elle est souvent confondue avec des diagnostics plus fréquents d’omphalocèle ou de gastroschisis.
Il est possible de soupçonner ces affections pendant la grossesse lorsque :
- La vessie n’est pas identifiée lors d’échographies successives (absence de remplissage vésical).
- Il existe une diminution de l’épaisseur de la paroi abdominale.
- Il existe une masse de la partie inférieure de l’abdomen, qui devient plus proéminente à mesure que la grossesse progresse.
- Il existe une insertion basse du cordon ombilical.
- Les organes génitaux ont une position anormale (antérieure ou postérieure) et il est difficile de déterminer le sexe du fœtus.
- Le phallus est court.
- Il existe une augmentation du diamètre pelvien, avec séparation des branches pubiennes.
- Il existe un omphalocèle.
- Il existe des anomalies des membres inférieurs et/ou un myéloméningocèle (évoquant une exstrophie cloaquale).
L’échographie tridimensionnelle et l’utilisation croissante de l’IRM fœtale amélioreront la capacité à diagnostiquer l’exstrophie vésicale et cloacale.21 Le diagnostic anténatal permet un conseil prénatal et l’organisation de l’accouchement dans un centre spécialisé dans l’exstrophie. Cela permet une approche multidisciplinaire par des équipes ayant l’expérience de la prise en charge du caractère unique du complexe exstrophie-épispadias.
Période néonatale
La plupart des variantes sont facilement identifiables à la naissance.
- L’exstrophie vésicale est le plus souvent observée chez les nouveau-nés à terme avec un bon poids de naissance.
- Les nouveau-nés atteints d’exstrophie cloacale sont habituellement prématurés et petits pour l’âge gestationnel.
Enfance
- Des variantes peu fréquentes peuvent passer inaperçues.
- Elles ne sont généralement identifiées plus tard dans la vie que par une incontinence urinaire persistante ou des troubles de la marche.
Examen physique
Dans la BEX classique, la plupart des anomalies sont des malformations de la paroi abdominale, de la vessie, des organes génitaux, des os du bassin et de l’anus, impliquant ainsi les voies urinaires inférieures, les organes génitaux et le système musculosquelettique (membres), tandis que dans l’exstrophie cloacale il existe une atteinte plus importante du tractus gastro-intestinal et du SNC.
Exstrophie vésicale classique
La paroi abdominale est allongée, et l’ombilic est bas situé, localisé sur le bord supérieur de la plaque vésicale ; il peut être associé à un défaut herniaire ou à une petite omphalocèle. La vessie est ouverte en avant, avec sa muqueuse entièrement exposée ; des polypes peuvent être visibles à sa surface. L’urine s’écoule par les orifices urétéraux à la surface vésicale. Une fermeture tardive peut entraîner des altérations inflammatoires ou mécaniques supplémentaires avec des signes d’inflammation muqueuse tels qu’un enduit blanchâtre, des ulcérations et des formations hyperplasiques. Les fines stries cutanées luisantes para-exstrophiques marquent la transition entre la peau normale et la zone de métaplasie malpighienne. L’anus est plus antérieur, mais la fonction sphinctérienne est normale. Les os du pubis sont largement séparés et peuvent aussi être raccourcis et en rotation externe (30 %). Ils sont palpables de part et d’autre de la plaque vésicale et à l’extrémité distale des bords triangulaires. Des hernies inguinales bilatérales sont palpables chez la plupart des patients des deux sexes.
- BEX féminin: il existe un clitoris bifide, des grandes lèvres séparées, et un mont du pubis divergent (Figure 2). Le vagin est plus court que la normale, d’une profondeur n’excédant pas 6 cm, mais de calibre normal. L’orifice vaginal est souvent sténosé et situé en position antérieure. Puisque l’anus est également en position ventrale, le périnée est raccourci.
- BEX masculin: Le défaut génital est sévère et est probablement l’un des aspects les plus problématiques de la reconstruction chirurgicale. La plaque urétrale est ouverte et s’étend le long d’un phallus court, large et incurvé dorsalement, depuis la vessie ouverte jusqu’au sillon glandulaire. Les deux corps caverneux sont situés sous la plaque urétrale. Un examen attentif met en évidence le colliculus séminal et les canaux éjaculateurs sous la forme de minuscules orifices dans la zone où la prostate est vraisemblablement située dorsalement. Le gland est ouvert et aplati (Figure 3), et les testicules de taille normale sont habituellement situés dans le scrotum.
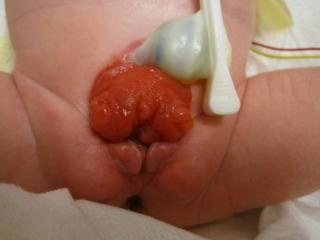
Figure 2 BEX classique avec un ombilic bas situé et un anus antériorisé dans le périnée. BEX féminine avec une vessie ouverte et une plaque urétrale, avec un clitoris bifide.
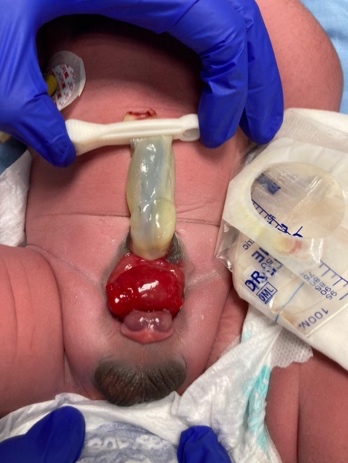
Figure 3 BEX masculin présentant un phallus court, large et à courbure dorsale. Distance anormale entre le scrotum et le pénis.
Épispadias
La malformation résulte d’un arrêt du développement se traduisant par la non-fermeture de la plaque urétrale et, en outre, par une localisation urétrale dorsale anormale. Ainsi, chez les garçons, on observe un méat ectopique ou une bande muqueuse sur le dos de la verge, et chez les filles, on met en évidence une fente urétrale de degré variable.8 La paroi abdominale et les muscles droits de l’abdomen, de même que l’ombilic, sont entièrement normalement développés. La symphyse pubienne est habituellement fermée, ou présente seulement un diastasis minime, indiquant des anomalies pelviennes et du plancher pelvien seulement mineures. L’incontinence urinaire semble être le principal symptôme clinique, en fonction du degré d’atteinte du sphincter urinaire. Dans la plupart des épispadias distaux, on n’observe pas de pertes urinaires involontaires, tandis que dans les formes proximales l’urine s’écoule en permanence par le méat. En raison de caractères cliniques parfois « mineurs », l’épispadias distal peut passer inaperçu à la naissance, en particulier chez les filles. Le diagnostic peut alors être posé plus tard, à l’âge scolaire, devant une incontinence urinaire résistante au traitement standard.
- **Épispadias masculin (Figure 4): Selon la localisation du méat, on distingue un épispadias pénopubien, pénien ou glandulaire.
- **Épispadias féminin (Figure 5): est divisé en trois degrés selon Davis, soit peu sévère avec un méat béant, intermédiaire, ou sévère avec une fente intéressant tout l’urètre et le col vésical, s’accompagnant en outre d’un prolapsus de la muqueuse vésicale.
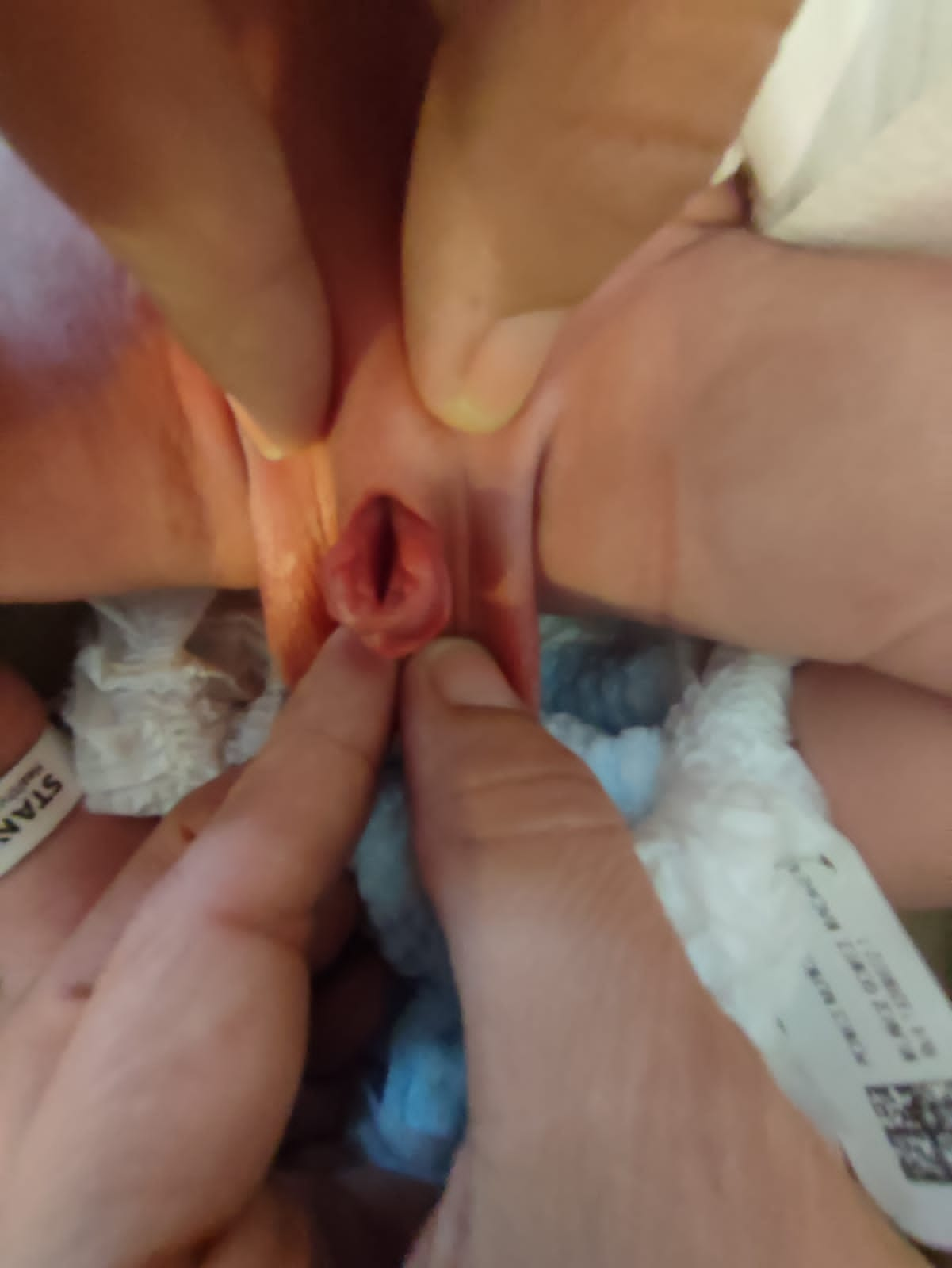
Figure 4 Épispadias masculin
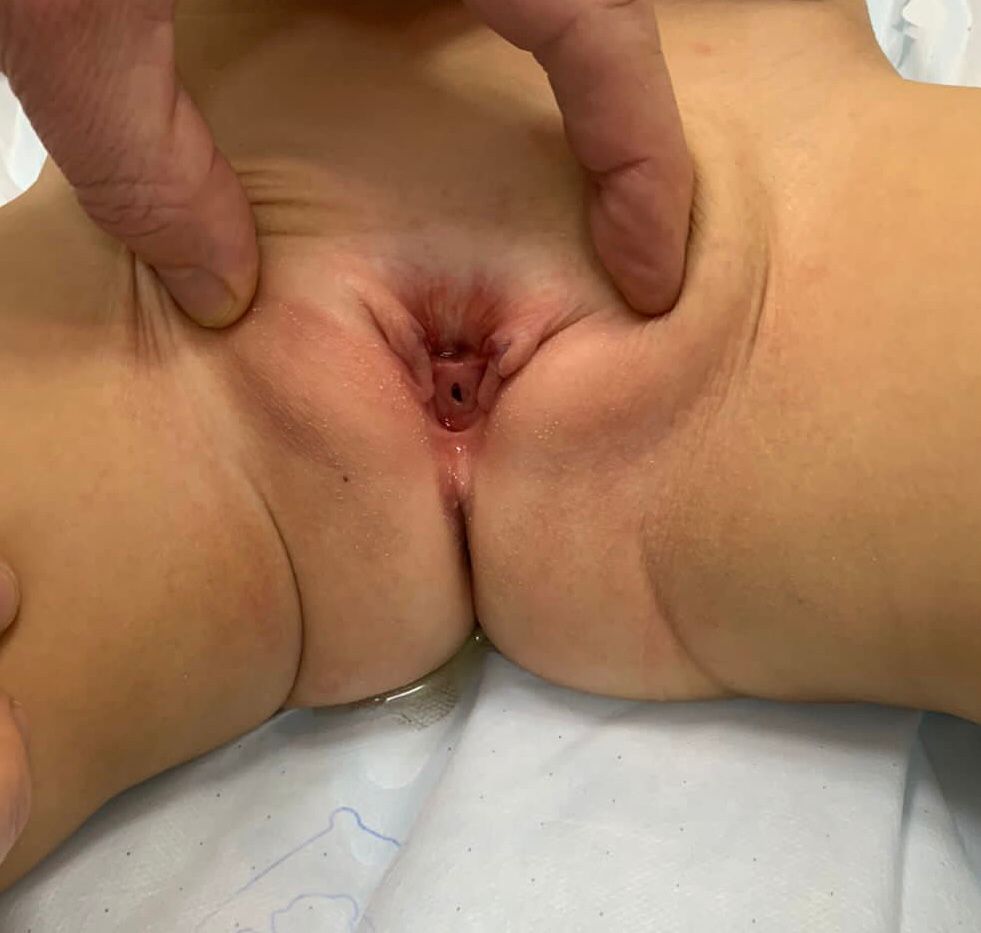
Figure 5 Épispadias féminin
Exstrophie cloacale
Les muscles droits de l’abdomen et les os pubiens sont séparés. La vessie est ouverte sur la paroi abdominale inférieure et divisée en deux moitiés adjacentes au segment exposé du cæcum. Les orifices qui mettent en communication l’iléon terminal, l’appendice (un ou deux) et l’intestin distal sont visibles au sein de la plaque cæcale, et l’iléon terminal peut faire procidence à travers celle-ci sous forme de « tronc » (aspect de trompe d’éléphant). Il s’accompagne d’un anus imperforé et peut être associé à un omphalocèle. 95 % présentent une myélodysplasie et 65 % une malformation des membres inférieurs.
-
CEX masculin: le phallus est généralement bifide et petit, chaque hémigland étant situé caudalement par rapport à chaque hémivessie, ou il peut être absent.
-
CEX chez la fille : le clitoris est bifide et il peut y avoir deux hémivagins avec un utérus bicorne (Figure 6).
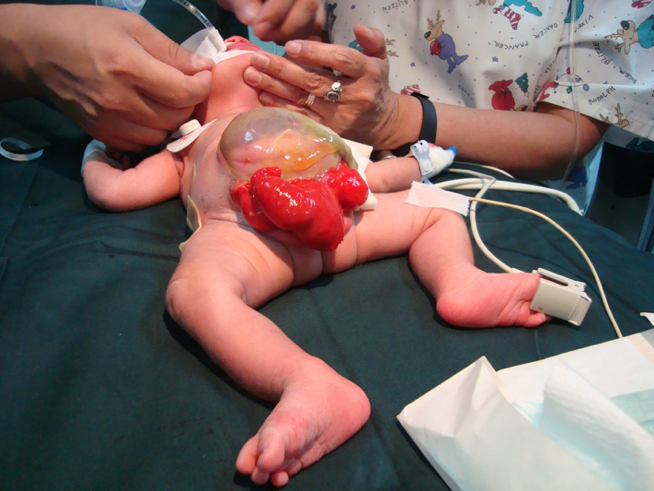
Figure 6 Exstrophie cloacale. Omphalocèle géante avec 2 hémivessies adjacentes à la plaque cæcale exstrophique. Phallus petit et bifide, avec son hémigland et un hémiscrotum situés distalement à chaque plaque vésicale.
Variantes de l’exstrophie
Cela comprend un spectre d’anomalies cliniquement vaste et hétérogène. L’exstrophie vésicale couverte apparaît similaire à l’exstrophie vésicale classique (BEX), avec une partie de la muqueuse vésicale recouverte de peau; elle peut également se présenter sous la forme d’un épispadias sévère avec une vessie prolabée. L’ombilic peut être en position orthotopique. La pseudo-exstrophie, toutefois, peut être très difficile à identifier après la naissance et est souvent diagnostiquée à un âge plus avancé. Les organes génitaux paraissent normaux, et les patients peuvent ne présenter aucun symptôme urinaire, certains étant complètement continents. À l’examen clinique, on peut retrouver un diastasis des grands droits de degré variable. Les radiographies du bassin peuvent mettre en évidence un diastasis de la symphyse pubienne, bien qu’il ne soit pas rare qu’il s’agisse d’une découverte fortuite.
Anomalies associées
Anomalies urologiques
Plusieurs malformations urologiques sont présentes chez environ un tiers de tous les cas d’EEC, principalement dans la population EC (obstruction de la jonction pyélo-urétérale, rein ectopique pelvien, rein en fer à cheval, agénésie rénale, mégauretère, ectopie urétérale et urétérocèle).1,8 Tous les patients devraient bénéficier d’une procédure antireflux lors de toute plastie du col vésical, car il existe une prévalence de 100 % de reflux vésico-urétéral bilatéral due à un défaut de développement de la jonction urétéro-vésicale dans l’ensemble du spectre de l’EEC.
Anomalies rachidiennes et orthopédiques
L’incidence des anomalies rachidiennes varie au sein du spectre de l’EEC. Chez les enfants nés avec CEB, des anomalies rachidiennes surviennent dans environ 7 % des cas, tandis qu’un groupe hétérogène d’anomalies rachidiennes congénitales résultant d’une fermeture défectueuse du tube neural tôt au cours de la vie fœtale et d’un développement anormal de la masse cellulaire caudale peut être confirmé par imagerie par résonance magnétique (IRM), identifié chez près de 100 % des patients CEX.8 Il faut garder à l’esprit une composante neurologique chez ces patients en ce qui concerne la fonction vésicale et celle des membres inférieurs, ainsi que la capacité érectile.22,23 Des anomalies squelettiques et des membres (déformations en pied bot, absence de pieds, déformations tibiales ou fibulaires et luxations de la hanche) sont plus fréquemment observées dans la CEX.1,8 Cependant, il n’existe pas de rapports sur la dysplasie de la hanche lors du suivi à long terme de l’EEC.
Anomalies gastro-intestinales
Celles-ci sont principalement associées au CEX. Outre un reliquat commun de l’intestin postérieur de taille variable, des omphalocèles sont retrouvées dans 88-100 % des cas de CEX. Une malrotation ou une duplication intestinales, ainsi que le syndrome de l’intestin court, peuvent être observés dans jusqu’à 46 % des cas.1,8 Dans environ 25 % des cas, un syndrome de l’intestin court, qu’il soit anatomique ou fonctionnel, entraîne un trouble de l’absorption.
Anomalies gynécologiques
Outre des organes génitaux externes anormaux, le col utérin s’insère bas au niveau de la paroi vaginale supérieure, à proximité de l’introïtus dans la plupart des cas.1,8,24 Néanmoins, l’anatomie et la fonction de l’utérus et des annexes utérines sont normales. Les anomalies du plancher pelvien et des muscles élévateurs de l’anus, associées à l’absence des ligaments cardinaux, prédisposent les femmes à un prolapsus vaginal ou utérin dans environ 50 % des cas. Les anomalies müllériennes sont assez fréquentes dans la CEX, c.-à-d. duplication du vagin et de l’utérus, agénésie vaginale.1,8
Examens d’imagerie
Ils sont généralement réalisés pour identifier et diagnostiquer d’éventuelles anomalies associées (malformations pelviennes et abdominales, anomalies de la colonne vertébrale et de la moelle épinière, etc).
Les principaux examens sont l’échographie rénale, l’échographie médullaire et les radiographies du rachis et du bassin. Tous les nouveau-nés atteints de CEX devraient bénéficier d’une échographie médullaire et de radiographies afin de définir les anomalies rachidiennes individuelles allant de l’hémivertèbre au myéloméningocèle. Une IRM est en outre recommandée au cours du suivi pour identifier des anomalies rachidiennes occultes prédisposant à un ancrage médullaire symptomatique.
L’évaluation échographique des articulations de la hanche est d’une importance fondamentale pour tous les patients atteints d’EEC. Une radiographie simple du bassin peut être utile pour estimer la dimension de l’écartement de la symphyse pubienne et la localisation des hanches.
Diagnostic
En anténatal, l’EEC peut être suspecté dès la 16e semaine de gestation. Avec les progrès de la sensibilité de l’échographie anténatale, la suspicion prénatale a augmenté.
Dans la plupart des cas, la malformation est diagnostiquée à la naissance, car elle devient cliniquement évidente.
Prise en charge
En cas de diagnostic anténatal ou de suspicion d’EEC, l’idéal est de conseiller les parents et l’accouchement doit être programmé dans un centre spécialisé (adresser si nécessaire). Il n’existe pas d’expérience étayant l’indication d’une chirurgie fœtale. L’accouchement par voie basse n’est pas recommandé en raison du risque accru de lésion de la plaque vésicale.
De nos jours, ce n’est plus considéré comme une urgence et/ou une urgence chirurgicale, ce qui permet un transfert dans des conditions de sécurité vers des centres spécialisés capables de prendre en charge ces enfants présentant des malformations rares et hautement complexes.
L’approche chirurgicale était initialement centrée sur la dérivation urinaire afin de préserver la fonction rénale. Cependant, depuis que la première fermeture vésicale réussie a été rapportée par Young en 1942,1,8,11,24,25 d’importants progrès ont été réalisés dans les domaines de la reconstruction en plusieurs temps, de l’aspect des organes génitaux externes et de la préservation de la continence et de la fonction rénale. Grâce aux améliorations des techniques chirurgicales, ainsi que des soins périopératoires, on peut désormais s’attendre à une excellente survie à long terme.
Chirurgie
Les objectifs de l’approche chirurgicale actuelle sont ;1 la reconstruction de la paroi abdominale,2 le repositionnement et la fermeture anatomique de la vessie exstrophiée,3 la préservation de la fonction rénale et l’obtention de la continence urinaire, et4 la reconstruction des organes génitaux externes.26,27,28,29,30 Historiquement, la reconstruction était réalisée en 3 étapes débutant en période néonatale. Cependant, il existe actuellement des centres ultra-spécialisés ou des équipes multicentriques qui prennent en charge cette affection et peuvent organiser une prise en charge différée, afin d’offrir le meilleur à chaque patient en accumulant de l’expérience dans des pathologies de faible fréquence et de grande complexité.31,32,33,34,35,36,37,38,39,40
Les étapes chirurgicales sont
- Fermeture de la vessie, avec rapprochement des os pubiens, sans nécessité d’ostéotomies. Chez les patients atteints de CEX, une dérivation intestinale doit être réalisée.
- La deuxième étape consiste en une reconstruction génitale complète. Dans certains centres, on réalise également la procédure de Kelly ou une reconstruction radicale des tissus mous, afin d’obtenir la continence vésicale et une plus grande longueur pénienne chez les hommes.
- Enfin, la continence urinaire doit être évaluée à moyen et à long terme. Pour les cas où cet objectif n’a pas encore été atteint ou lorsqu’il existe un risque d’altération de la fonction rénale, il existe une troisième étape au cours de laquelle une chirurgie du col vésical et/ou une cystoplastie d’augmentation peuvent être réalisées, avec une dérivation urinaire continente permettant le cathétérisme intermittent propre.
Dans divers centres surspécialisés à travers le monde, la fermeture différée à l’âge de deux à quatre mois a été privilégiée, en réalisant une approche en un seul temps, associée à la chirurgie décrite par Kelly,26,30,39 qui comprend:1 la dissection et la fermeture de la vessie avec ou sans procédure antireflux (si la taille de la vessie et les caractéristiques de sa paroi le permettent);2 la mobilisation radicale des tissus mous avec désinsertion des branches pubiennes;3 la création d’un col vésical ou d’un mécanisme de continence à l’aide du stimulateur de Peña pour identifier le néo-complexe musculaire sphinctérien et4 la reconstruction pénienne ou l’introitoplastie.
Les avantages de cette approche sont qu’une chirurgie pendant la période néonatale est évitée, permettant de préserver les tissus natifs et d’éviter les cicatrices et la fibrose générées par une fermeture primaire. Cela favorise également l’attachement parental et le développement d’un lien entre l’enfant et ses parents, sans nécessiter une hospitalisation prolongée au cours des premiers jours de vie.
Il a été démontré que la fermeture vésicale différée peut être réalisée en toute sécurité et avec succès, et qu’en principe elle n’affecte pas le développement vésical. Le report de l’intervention permet la maturation du nouveau-né, avec à la clé une réduction des risques liés à une anesthésie prolongée, et favorise la croissance du nourrisson pendant la période de minipuberté, en plus de favoriser la préparation de l’équipe d’experts pour chaque cas.39
Complications
En raison de leur complexité et de leur très faible fréquence, ces affections présentent une série de complications principalement associées à la chirurgie reconstructrice:34,39
- Précoces : déhiscence de plaie chirurgicale, prolapsus vésical, fistules urétrales ou vésico-cutanées (4 – 19 %), sténose urétrale (8 %), ischémie pénienne.
- Tardives : RVU, infections urinaires récidivantes, vidange vésicale incomplète, incontinence urinaire, lithiase vésicale, fibrose de la paroi abdominale, rupture vésicale, prolapsus utérin, insuffisance rénale, éjaculation rétrograde, oligospermie et infertilité, ainsi que le syndrome de l’intestin court et l’incontinence fécale (exstrophie cloacale).
Pronostic
Si ces patients ne sont pas traités, ils souffriront d’infections urinaires récidivantes, d’une incontinence urinaire permanente et de troubles de la fonction sexuelle, avec également un risque accru de cancer de la vessie, ce qui, pris ensemble, se traduit par une qualité de vie moindre.26,27,28,29,30,31,32,33,34,35
Cependant, si une prise en charge appropriée de la pathologie est effectuée comme suggéré, ces patients ont une qualité de vie attendue probablement similaire à celle de la population générale. Il est également estimé qu’ils pourront potentiellement mener une vie absolument normale, étant donné que dans plus de 90% des cas il n’existe pas d’association avec d’autres malformations congénitales. En outre, ces patients pourraient acquérir une continence urinaire sans nécessiter une augmentation vésicale ni un cathétérisme intermittent pour la vidange, même si le taux de succès reste réservé (seulement 23% de mictions volontaires par l’urètre avec la technique MSRE,41 39% avec la technique CPRE,28 et 53% avec la technique de Kelly26).
Tableau 1 Large éventail des taux de continence des différentes approches en fonction de la définition de la continence et de la période d’observation.42
| Approche | Taux de continence (%) | Littérature |
|---|---|---|
| MSRE | 74 | Gearhart et al |
| 62 | Gupta et al | |
| 22 | Dickson et al | |
| CPRE | 80 | Grady et al |
| 74 | Hammouda et al | |
| 23 | Arab et al | |
| RSTM | 73 | Kelly et al |
| 70 | Jarzebowski et al | |
| 33–67 (femme) | Cuckow et al | |
| 44–81 (homme) |
Actuellement, ces patients présentent une excellente survie à long terme, avec un taux de continence urinaire allant jusqu’à 75-80 % chez les patients atteints d’exstrophie vésicale et 65-70 % dans les cas d’exstrophie cloaquale;39,43,41 cependant, beaucoup d’entre eux nécessitent des dérivations urinaires et intestinales permanentes. En général, la fonction sexuelle est préservée, et la plupart des patients sont fertiles. L’accouchement par césarienne est recommandé chez les patientes opérées afin d’éviter d’endommager les mécanismes de la continence.
Références
- Gearhart JP, Gearhart JP, Rink RC. The Bladder Exstrophy–epispadias–cloacal Exstrophy Complex. J Pediatr Urol 2001: 386–415. DOI: 10.1016/b978-1-4160-3204-5.00030-x.
- Siffel C, Correa A, Amar E, Bakker MK, Bermejo-Sánchez E, Bianca S, et al.. Bladder exstrophy: An epidemiologic study from the International Clearinghouse for Birth Defects Surveillance and Research, and an overview of the literature. Am J Med Genet C Semin Med Genet 2011; 157 (4): 321–332. DOI: 10.1002/ajmg.c.30316.
- Cervellione RM, Mantovani A, Gearhart J, Bogaert G, Gobet R, Caione P, et al.. Prospective study on the incidence of bladder/cloacal exstrophy and epispadias in Europe. J Pediatr Urol 2015; 11 (6): 337.e1–337.e6. DOI: 10.1016/j.jpurol.2015.03.023.
- Boyadjiev SA, Dodson JL, Radford CL, Ashrafi GH, Beaty TH, Mathews RI, et al.. Clinical and molecular characterization of the bladder exstrophy-epispadias complex: analysis of 232 families. BJU Int. 2004;94:1337-1343. . DOI: 10.1111/j.1464-410X.2004.05170.x..
- Männer J, Kluth D. The morphogenesis of the exstrophy-epispadias complex: a new concept based on observations made in early embryonic cases of cloacal exstrophy. Anat Embryol (Berl) 2005; 210 (1): 51–57. DOI: 10.1007/s00429-005-0008-6.
- Marshall VF, Muecke EC. Variations in exstrophy of the bladder. Plast Reconstr Surg 1962; 31 (4): 396. DOI: 10.1097/00006534-196304000-00030.
- Stephens FD, Hutson JM. Differences in embryogenesis of epispadias, exstrophy–epispadias complex and hypospadias. J Pediatr Urol 2005; 1 (4): 283–288. DOI: 10.1016/j.jpurol.2005.01.008.
- Ebert AK, Reutter H, Ludwig M, Rösch WH. Exstrophy-epispadias complex. Definitions 2009; 4 (23). DOI: 10.32388/7eqi40.
- Ambrose SS, O’Brien DP. Surgical Embryology of the Exstrophy-Epispadias Complex. Surg Clin North Am 1974; 54 (6): 1379–1390. DOI: 10.1016/s0039-6109(16)40493-7.
- Mildenberger H, Kluth D, Dziuba M. Embryology of bladder exstrophy. J Pediatr Surg. 1988;23:166-170. . DOI: 10.1016/s0022-3468(88)80150-7..
- Gearhart JP. Exstrophy-Epispadias Complex. Campbell-Walsh-Wein Urology, vol. 31. 12th ed. 2021. DOI: 10.1016/b978-1-4160-6911-9.00124-9.
- Ives E, Coffey R, Carter CO. A family study of bladder exstrophy. J Med Genet. 1980;17:139-141. . DOI: 10.1136/jmg.17.2.139..
- Shapiro E, Lepor H, Jeffs RD. The inheritance of the exstrophy-epispadias complex. J Urol. 1984;132:308-310. .
- Reutter H, Qi L, Gearhart JP, Boemers T, Ebert AK, Rösch WH, et al.. Concordance analyses of twins with bladder exstrophy-epispadias complex suggest genetic etiology. Am J Med Genet A. 2007;143:2751-2756. . DOI: 10.1002/ajmg.a.31975.
- Carter CO. Genetics of common single malformations. Br Med Bull. 1976;32:21-26. .
- Ludwig M, Ching B, Reutter H, Boyadjiev SA. The bladder exstrophy-epispadias complex. Birth Defects Res Part A Clin Mol Teratol. 2009;85:509-22. . DOI: 10.1002/bdra.20557..
- Reutter H, Shapiro E, Gruen JR. Seven new cases of familial isolated bladder exstrophy and epispadias complex (BEEC) and review of the literature. Am J Med Genet A. 2003;120A:215-221. . DOI: 10.1002/ajmg.a.20057..
- Gambhir L, Höller T, Müller M, Schott G, Vogt H, Detlefsen B, et al.. Epidemiological survey of 214 European families with Bladder Exstrophy-Epispadias Complex (BEEC) J Urol. 2008;179:1539-1543. . DOI: 10.1016/j.juro.2007.11.092..
- Byron-Scott R, Haan E, Chan A, Bower C, Scott H, Clark K. A population-based study of abdominal wall defects in South Australia and Western Australia. Paediatr Perinat Epidemiol. 1998;12:136-151. . DOI: 10.1046/j.1365-3016.1998.00090.x..
- Wood HM, Babineau D, Gearhart JP. In vitro fertilization and the cloacal/bladder exstrophy-epispadias complex: A continuing association. J Pediatr Urol. 2007;3:305-310. . DOI: 10.1016/j.jpurol.2006.10.007..
- Palmer B, Frimberger D, Kropp B. Bladder exstrophy-epispadias complex and cloacal exstrophy. Section 7.D: Developmental anomalies. Paediatric Urology Book. Paediatric Urology Department, University of Oklahoma; 2011. DOI: 10.1007/978-1-84882-132-3\\_45.
- Rösch WH, Hanisch E, Hagemann M, Neuhuber WL. The Characteristic innervation pattern of the urinary bladder in particular forms of exstrophy-epispadias-complex. BJU. 2001;87:30. .
- Schober JM, Carmichael PA, Hines M, Ransley PG. The ultimate challenge of cloacal exstrophy. J Urol. 2002;167:300-304. . DOI: 10.1016/s0022-5347(05)65455-9..
- Woodhouse CRJ, Hinsch R. The anatomy and reconstruction of the adult female genitalia in classical exstrophy. BJU. 1997;79:618-622. . DOI: 10.1046/j.1464-410X.1997.00148.x..
- Woodhouse. C.R.J.: Genitoplasty in exstrophy and epispadias. Cambridge University Press; 2006, DOI: 10.1017/cbo9780511545757.046.
- Kelly JH. Vesical exstrophy: repair using radical mobilisation of soft tissues. Pediatr Surg Int 1995; 10 (5-6): 298–304. DOI: 10.1007/bf00182207.
- Grady RW, Mitchell ME. Complete primary repair of exstrophy. J Urol. 1999; 62 (4): 415–1420. DOI: 10.1097/00005392-199910000-00071.
- Groth. Bladder exstrophy consortium (MIBEC) after 5 years. AUA Chicago; 2019.
- Dickson AP. The management of bladder exstrophy: the Manchester experience. J Pediatr Surg. 2014; 9 (2): 44–250. DOI: 10.1016/j.jpedsurg.2013.11.031.
- Cuckow P, López PJ. Bladder Exstrophy Closure and Epispadias. In: Spitz L, Coran A, editors. En Operative Pediatric Surgery. 7th ed. 2013. DOI: 10.1201/b13237-101.
- Borer JG, Gargollo PC, Hendren WH, Diamond DA, Peters CA, Atala A, et al.. Early Outcome Following Complete Primary Repair Of Bladder Exstrophy In The Newborn. J Urol 2005; 174 (4 Part 2): 1674–1679. DOI: 10.1097/01.ju.0000175942.27201.59.
- Baird AD, Nelson CP, Gearhart JP. Modern staged repair of bladder exstrophy: a contemporary series. J Pediatr Urol 2007. 4: 11–315. DOI: 10.1016/j.jpurol.2006.09.009.
- Borer JG, Vasquez E, Canning DA, Kryger JV, Mitchell ME. An initial report of a novel multi-institutional bladder exstrophy consortium: a collaboration focused on primary surgery and subsequent care. J Urol. 2015. DOI: 10.1016/j.juro.2014.10.114.
- Ellison JS, Shnorhavorian M, Willihnganz-Lawson K, Grady R, Merguerian PA. A critical appraisal of continence in bladder exstrophy: Long-term outcomes of the complete primary repair. J Pediatr Urol 2016. 2 (4): 05 1–205 2057. DOI: 10.1016/j.jpurol.2016.04.005.
- Schaeffer AJ, Stec AA, Purves JT, Cervellione RM, Nelson CP, Gearhart JP. Complete primary repair of bladder exstrophy: a single institution referral experience. J Urol. 2011; 86 (3): 041–1046. DOI: 10.1016/j.juro.2011.04.099.
- Pathak P, Ring JD, Delfino KR, Dynda DI, Mathews RI. Complete primary repair of bladder exstrophy: a systematic review. J Pediatr Urol. 2020; 6 (2): 49–153. DOI: 10.1016/j.jpurol.2020.01.004.
- Ahn JJ, Shnorhavorian M, Katz C, Goldin AB, Merguerian PA. Early versus delayed closure of bladder exstrophy: A National Surgical Quality Improvement Program Pediatric analysis. J Pediatr Urol. 2018; 4 (1): 7 1–27 5. DOI: 10.1016/j.jpurol.2017.11.008.
- Leclair MD, Villemagne T, Faraj S, Suply E. The radical soft-tissue mobilization (Kelly repair) for bladder exstrophy. J Pediatr Urol. 2015; 1 (6): 64–365. DOI: 10.1016/j.jpurol.2015.08.007.
- Leclair MD, Faraj S, Sultan S. One-stage combined delayed bladder closure with Kelly radical soft-tissue mobilization in bladder exstrophy: preliminary results. J Pediatr Urol. 2018; 4 (6): 58–564. DOI: 10.1016/j.jpurol.2018.07.013.
- Baradaran N, Stec AA, Schaeffer AJ, Gearhart JP, Mathews RI. Delayed primary closure of bladder exstrophy: immediate postoperative management leading to successful outcomes. Urology. 2012; 9 (2): 15–419. DOI: 10.1016/j.urology.2011.08.077.
- Jarzebowski AC, McMullin ND, Grover S SR, BR H, J.M.. The Kelly technique of bladder exstrophy repair: continence, cosmesis and pelvic organ prolapse outcomes. J Urol. 2009. DOI: 10.1016/j.juro.2009.02.083.
- Cervellione RM, Husmann DA, Bivalacqua TJ, Sponseller PD, Gearhart JP. Penile ischemic injury in the exstrophy/epispadias spectrum: new insights and possible mechanisms. J Pediatr Urol. 2010; 5: 50–456. DOI: 10.1016/j.jpurol.2010.04.007.
- Purves JT, Gearhart JP. Complications of radical soft-tissue mobilization procedure as a primary closure of exstrophy. J Pediatr Urol 2008. 1: 5–69. DOI: 10.1016/j.jpurol.2007.02.006.
- Maruf M, Manyevitch R, Michaud J. Urinary Continence Outcomes in Classic Bladder Exstrophy: A Long-Term Perspective. J Urol. 2020; 03 (1): 00–205. DOI: 10.1097/ju.0000000000000505.
- Promm M, Roesch WH. Recent Trends in the Management of Bladder Exstrophy: The Gordian Knot Has Not Yet Been Cut. Front Pediatr 2019; 7. DOI: 10.3389/fped.2019.00110.
Dernière mise à jour: 2025-09-22 07:59