
12: Anomalías congénitas del riñón y de las vías urinarias
Este capítulo durará aproximadamente 15 minutos para leer.
Introducción
Las anomalías congénitas del riñón y de las vías urinarias (CAKUT) son un grupo de anomalías estructurales que afectan a los riñones y las vías urinarias. Se estima que hay aproximadamente de 4 a 60 casos por cada 10,000 nacimientos en la población general. El diagnóstico de CAKUT suele realizarse durante una ecografía prenatal de rutina o en el periodo posnatal en un recién nacido con signos y síntomas clínicos asociados.1 La prevalencia de CAKUT en los lactantes pretérmino fue del 2% en una cohorte reciente, de gran tamaño y publicada, con 409704 lactantes.2
La anomalía más común es la obstrucción de la unión pieloureteral, que afecta aproximadamente al 20% de las personas con la afección. Entre los otros tipos de CAKUT, se incluyen los riñones multiquísticos displásicos, la agenesia renal, la displasia renal, la hipoplasia renal, el reflujo vesicoureteral, el megauréter, el sistema colector doble, el uréter ectópico y las válvulas uretrales posteriores (Figura 1).
A pesar de los avances significativos logrados en la detección y el diagnóstico de CAKUT, las causas subyacentes de la afección aún no se comprenden por completo. Se necesita más investigación para mejorar nuestra comprensión de los factores embriológicos, genéticos y ambientales que contribuyen al desarrollo de CAKUT y para comprender mejor sus causas subyacentes y mejorar las estrategias de prevención y tratamiento.
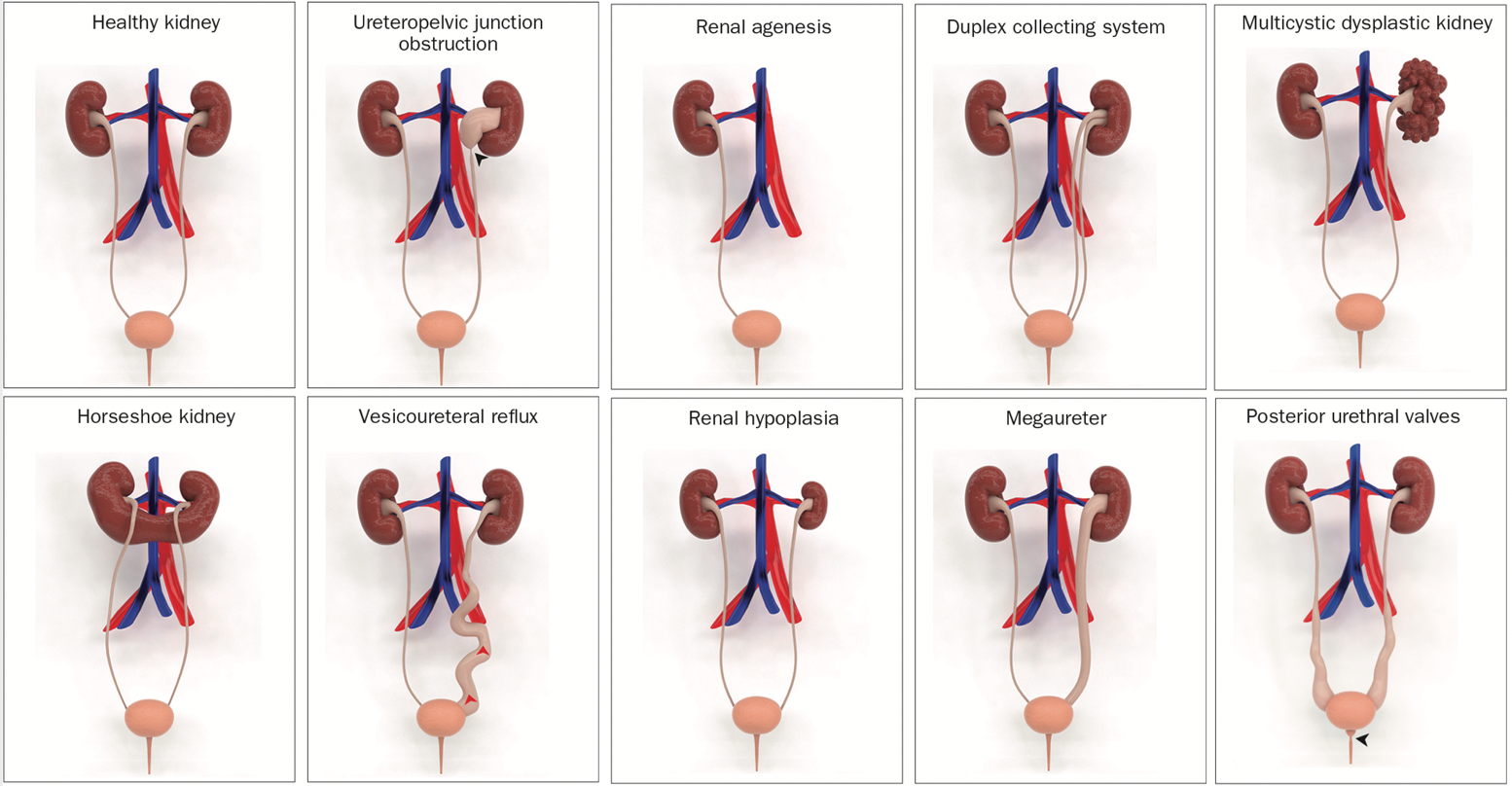
Figura 1 Modelos 3D ilustrativos de anomalías congénitas del riñón y del tracto urinario (CAKUT).3
Embriología
El desarrollo de los riñones y del tracto urinario en el feto implica un proceso complejo que comienza en las primeras semanas del embarazo e incluye la formación y diferenciación de varias estructuras. Este proceso se inicia con la formación de las yemas ureterales, que surgen del mesodermo intermedio y dan origen a los túbulos colectores y, en última instancia, a los riñones. Los riñones también se desarrollan a partir de una estructura llamada metanefros, que se forma a partir del mesodermo en el embrión.
Durante el desarrollo, las yemas ureterales y el metanefros deben conectarse y diferenciarse adecuadamente para formar los riñones funcionales y las vías urinarias. Las alteraciones en este proceso pueden dar lugar a una variedad de anomalías congénitas.
CAKUT puede clasificarse por localización (riñones, uréteres, vejiga o uretra), por causa (mutaciones genéticas, exposiciones ambientales, infecciones durante el embarazo, etc.), por patrón de herencia y por gravedad (Tabla 1). En el 10% al 25% de los casos, CAKUT se atribuye a trastornos genéticos. CAKUT presenta manifestaciones fenotípicas complejas en asociación con trastornos genéticos (Figura 2 y Figura 3).4 Es posible que individuos con las mismas anomalías del genoma se manifiesten con diferentes formas de CAKUT, y anomalías fenotípicas similares pueden surgir debido a distintos trastornos genéticos, y asociarse con enfermedades extrarrenales.2
Tabla 1 Categorías de los trastornos CAKUT
| Riñón | Sistema colector |
|---|---|
| Número: agenesia renal | Obstrucción de la unión ureteropélvica |
| Morfología: hipoplasia renal, displasia, MCDK | Reflujo vesicoureteral |
| Posición: riñón en herradura, ectópico, pélvico | Megauréter |
| Sistema dúplex | |
| VUP |
Varias malformaciones del parénquima renal dan lugar a un fallo del desarrollo normal de las nefronas, entre ellas la displasia quística, la displasia renal, la agenesia renal, la disgenesia tubular renal y la disgenesia tubular renal.
Enfermedad renal poliquística
El término enfermedad renal poliquística se utiliza generalmente para describir una enfermedad en la que hay múltiples quistes en los riñones sin displasia asociada.
La enfermedad renal poliquística autosómica dominante (ADPKD) y la enfermedad renal poliquística autosómica recesiva (ARPKD) son los dos tipos más comunes de enfermedad renal poliquística. Además del patrón de herencia, ya sea dominante o recesivo, la ARPKD y la ADPKD difieren en cuanto a sus manifestaciones clínicas y pronósticos. La ADPKD es la forma más común de PKD y está causada por mutaciones en los genes PKD1 o PKD2.
Genes implicados en el desarrollo de la enfermedad quística renal:5,6
- PKD1: Este gen está asociado con la ADPKD. Las mutaciones en el gen PKD1 son responsables de aproximadamente el 85% de los casos de ADPKD.
- PKD2: Este gen también está asociado con la ADPKD. Las mutaciones en el gen PKD2 son responsables de aproximadamente el 15% de los casos de ADPKD.
- HNF1β: Las mutaciones en este gen están asociadas con una poliquistosis renal autosómica dominante tipo 2 (ADPKD2).
- REN: Este gen está asociado con una poliquistosis renal infantil.
- MUC1: Las mutaciones en este gen están asociadas con una enfermedad renal quística medular relacionada con MUC1.
- INVS: Las mutaciones en este gen están asociadas con una enfermedad renal quística medular relacionada con nefronoftisis.
- NPHP1, NPHP3, NPHP4, NPHP5, NPHP6, NPHP7, NPHP8, NPHP9: Estos genes están asociados con una enfermedad renal quística medular relacionada con nefronoftisis.
- BMP7: Las mutaciones en este gen están asociadas con una enfermedad renal quística medular relacionada con nefronoftisis
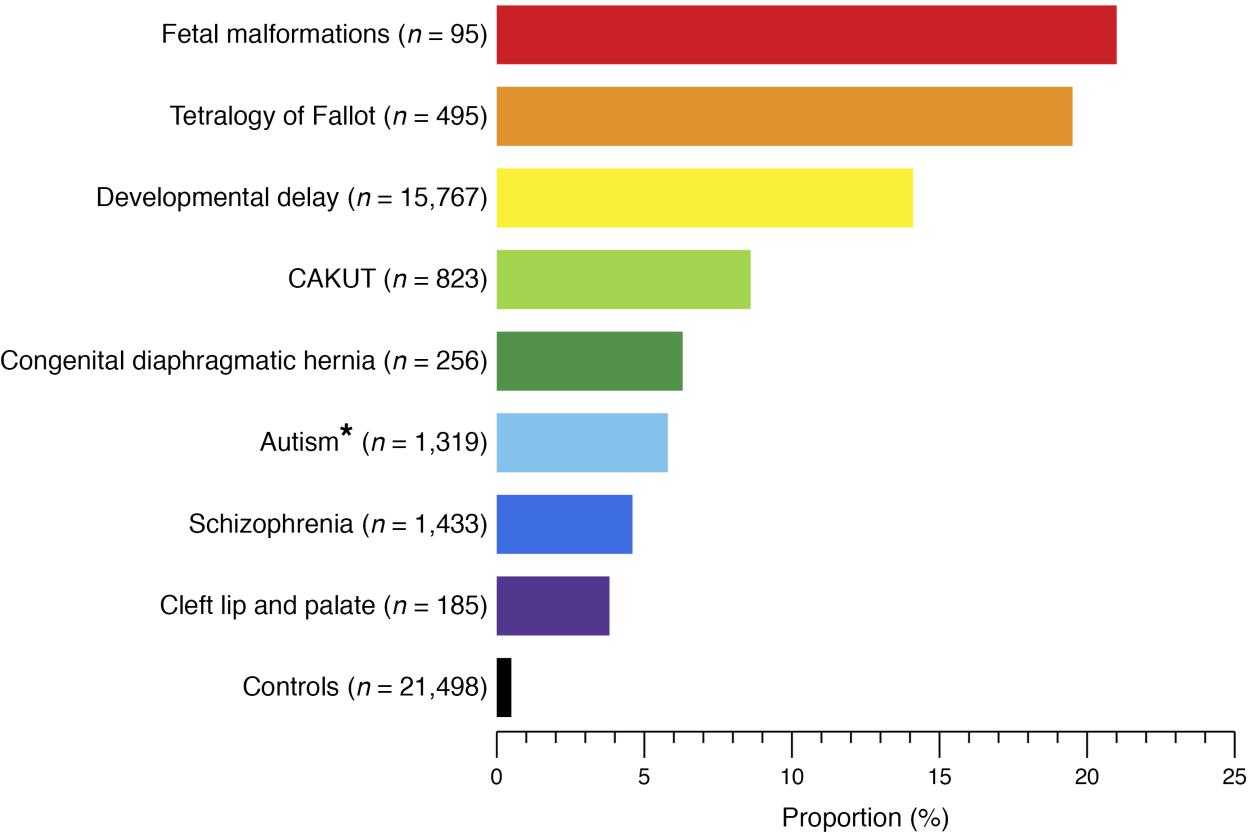
Figura 2 Proporción de pacientes con trastornos genómicos conocidos en diferentes fenotipos del desarrollo humano y controles sanos.4
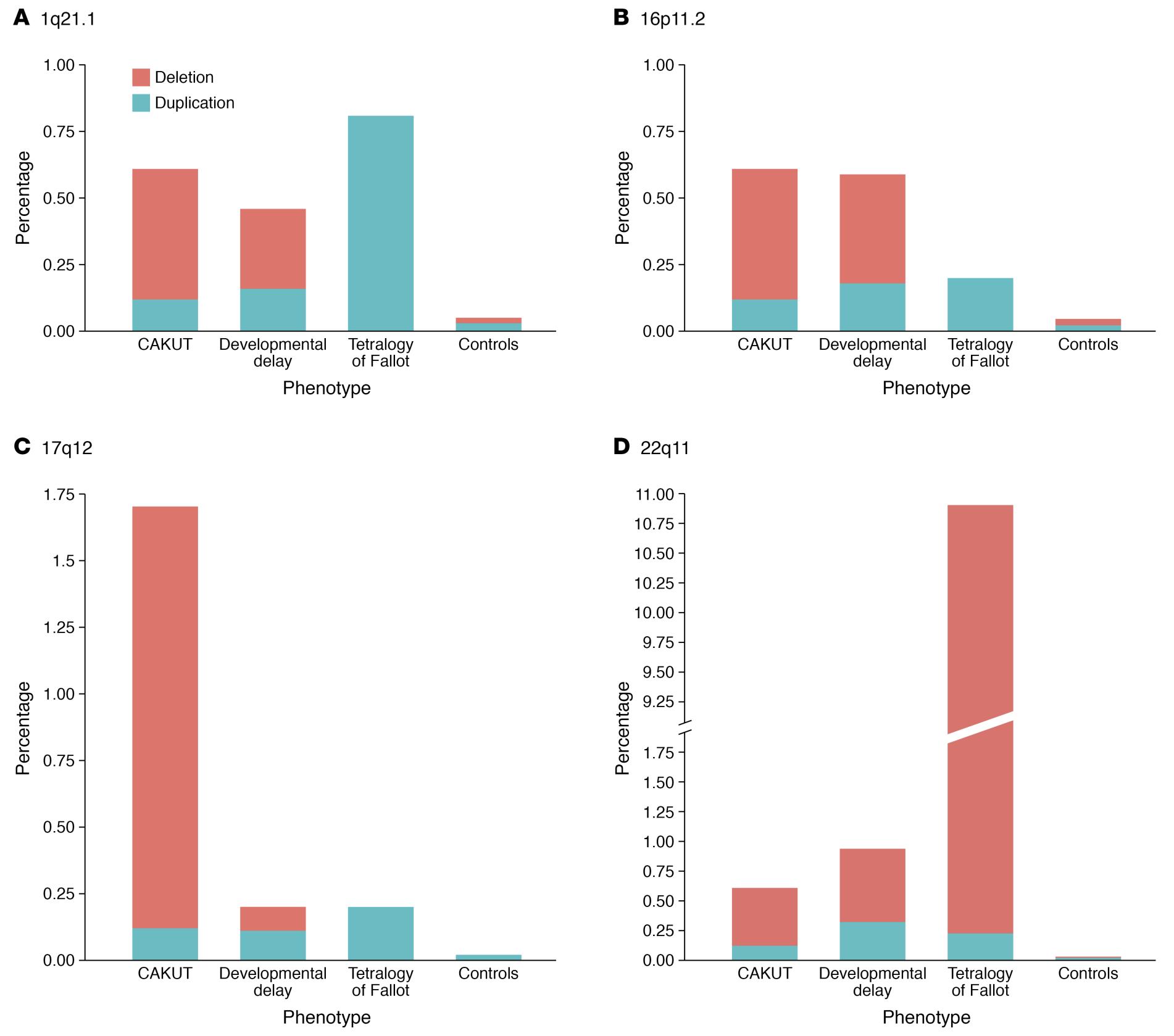
Figura 3 Diferencias y similitudes en la prevalencia de los cuatro loci de CNV implicados con mayor frecuencia en pacientes con CAKUT.4
MCDK
El MCDK es una enfermedad quística congénita no hereditaria, un tipo de displasia renal que causa múltiples quistes no comunicantes separados por parénquima displásico. La prevalencia de MCDK es de aproximadamente 1 en 3,600 a 1 en 4,300 nacidos vivos.7,8 La causa del MCDK es desconocida, pero se piensa que podría estar relacionada con una formación anómala del brote ureteral, la exposición a teratógenos o la obstrucción del tracto urinario. El MCDK suele detectarse mediante ecografía prenatal. Como a menudo no causa síntomas ni complicaciones, por lo general solo se identifica posnatalmente si hay una masa evidente o si se descubre de forma incidental durante estudios de imagen realizados por otra afección.
Algunos de los genes que se han asociado con MCDK incluyen:9,10,11
- HNF1β: Este gen codifica una proteína llamada factor nuclear hepatocitario 1 beta, que desempeña un papel en el desarrollo y la función del riñón y de otros órganos. Las mutaciones en HNF1β pueden causar una gama de anomalías renales, incluida MCDK.
- PAX2: Este gen codifica una proteína llamada caja pareada 2, que es importante para el desarrollo del uréter, el conducto que transporta la orina desde el riñón hasta la vejiga. Las mutaciones en PAX2 pueden causar MCDK y otras anomalías renales.
- SIX1: Este gen codifica una proteína llamada sine oculis homeobox 1, que participa en el desarrollo de varios órganos, incluido el riñón. Las mutaciones en SIX1 pueden causar MCDK y otras anomalías renales.
- WT1: Este gen codifica una proteína llamada tumor de Wilms 1, que es importante para el desarrollo del riñón y de otros órganos. Las mutaciones en WT1 pueden causar MCDK y otras anomalías renales.
El MCDK se maneja típicamente de forma conservadora (es decir, con observación), ya que la mayoría de los casos no presentan complicaciones a largo plazo. Los pacientes con anomalías contralaterales pueden presentar deterioro de la función renal con el tiempo. Por lo tanto, es necesario un seguimiento a largo plazo.
Agenesia renal
En comparación con la agenesia renal bilateral, la agenesia renal unilateral ocurre en uno de cada 1000 embarazos y tiene un pronóstico favorable. Se estima que entre el 0,1% y el 0,3% de 1.000 nacimientos se ven afectados por agenesia renal bilateral, lo que generalmente resulta en tasas de supervivencia bajas. Se han descrito varios patrones de herencia en familias con agenesia renal, que puede presentarse como un hallazgo aislado o como parte de un síndrome.12
La agenesia renal unilateral puede ser causada por mutaciones en varios genes, incluidos ATRX, RET, BMP4, FRAS1, FREM1, GRIP1, GLI3, UPK3A, entre otros. Además, la agenesia renal bilateral puede ser causada por mutaciones en los genes RET, FGF20, ITGA8. Genes relacionados enumerados en la Tabla 2.13,14
Displasia renal
La hipodisplasia renal resulta de una detención del desarrollo durante el primer trimestre de la vida fetal del blastema renal metanéfrico. Sin embargo, se desconoce qué causa la hipodisplasia renal; las anomalías vasculares in útero, así como los trastornos del desarrollo de origen genético, son dos mecanismos posibles. La hipodisplasia renal puede ser un hallazgo aislado o formar parte de un síndrome genético asociado con malformaciones del tracto urinario. El Estudio ESCAPE proporcionó un análisis de genes del desarrollo (HNF1β, PAX2, EYA1, SIX1 y SALL1) implicados en la displasia renal en una gran cohorte de niños con hipodisplasia renal. Según este estudio, se diagnosticaron mutaciones en PAX2 en el 15% de los pacientes con patología renal, y aquellos con deleciones genéticas o mutaciones de genes de gran tamaño mostraron fenotipos renales variables. Además, el 22% de todos los pacientes con hipodisplasia renal quística presentaron una mutación en HNF1, lo que sugiere que a los individuos con displasia renal quística se les realice cribado de mutaciones en HNF1.15 Los hallazgos en la ecografía renal que muestran reducción del tamaño renal y pérdida de la diferenciación corticomedular son indicativos clínicos de hipodisplasia renal. Con el tiempo, algunos pacientes con hipodisplasia renal pueden desarrollar enfermedad renal terminal. No existe un tratamiento específico para la hipodisplasia renal. Es importante el seguimiento y manejo de la función renal durante años. Para los pacientes que desarrollan enfermedad renal terminal, el trasplante renal es la opción preferida.
Unión pieloureteral
La causa patológica más común de la hidronefrosis neonatal es la obstrucción de la parte superior del uréter. La incidencia global es de 1:1,500, con una relación de 2:1 en recién nacidos varones respecto a mujeres.16 Una obstrucción de la UPJ suele deberse a una estenosis intrínseca o, con menor frecuencia, a un vaso cruzado que provoca compresión extrínseca en la parte superior del uréter. Un posible mecanismo es que exista una alteración embriológica en el uréter proximal que modifique el desarrollo de la musculatura circular, al alterar las fibras de colágeno y la composición entre y alrededor de las células musculares.
Varias mutaciones de un solo gen están implicadas en la UPJO, incluidas Id2, PAX2, EYA, AGTR2, BMP4, SOX17, CHD1L y DSTYK.17,18 Se ha informado que Id2 y Adamts1 producen un fenotipo similar al fenotipo humano de la UPJO. Los ratones carentes de Id2 también presentan una inserción ureteral alta en la pelvis renal.19
Reflujo vesicoureteral
Se han identificado varios genes asociados con el desarrollo del VUR. Algunos de estos genes participan en el desarrollo y la función del músculo liso ureteral. Otros genes participan en el desarrollo y la función de la vejiga. La alta prevalencia de VUR entre gemelos, hermanos y descendencia sugiere una base genética para esta enfermedad. Se ha establecido que el VUR es una enfermedad genéticamente heterogénea con base en los resultados de estudios genéticos en diversas poblaciones. Un estudio de ligamiento y asociación a escala del genoma vinculó el VUR primario no sindrómico con la región 10q26 del genoma.20,21 Otros genes que se han asociado con el desarrollo del VUR incluyen el gen FOXC1, el gen MYH11 y el gen ACTN4. Estos genes desempeñan diversas funciones en el desarrollo y la función del tracto urinario.22,23 Un estudio de asociación de todo el genoma que utiliza el mayor análisis de variantes en el número de copias y estudio de asociación de todo el genoma de VUR está resultando en la identificación de cinco loci que podrían estar asociados con el VUR, incluidos WDPCP, OTX1, BMP5, WDPCP y WNT5, con efectos de gran magnitud. WNT5 puede desempeñar un papel en el desarrollo del sistema urogenital. Un estudio de asociación de todo el genoma que utiliza el mayor análisis de variantes en el número de copias y estudio de asociación de todo el genoma de VUR está resultando en la identificación de cinco loci que podrían estar asociados con el VUR, incluidos WDPCP, OTX1, BMP5, WDPCP y WNT5, con efectos de gran magnitud. WNT5 puede desempeñar un papel en el desarrollo del sistema urogenital.24
Válvulas uretrales posteriores
La válvula uretral posterior es una anomalía congénita del tracto urinario potencialmente mortal que suele descubrirse durante el periodo neonatal y que, a pesar del tratamiento óptimo, resulta en tasas elevadas de insuficiencia renal. Se estima que la PUV ocurre en 1 de cada 7,000–8,000 nacidos vivos.25
Existe una amplia gama de informes que indican que la PUV es la causa principal de enfermedad renal crónica en recién nacidos varones, y la ESRD puede presentarse en un rango del 5% al 64% de los casos.26,27 Una revisión sistemática informó que la enfermedad renal crónica (CKD) y la enfermedad renal terminal (ESKD) están asociadas con los pacientes con PUV con un riesgo de hasta 32% y 20%, respectivamente.28
Hasta la fecha, se han identificado pocos genes asociados con las válvulas uretrales posteriores. El síndrome de Prune Belly se ha vinculado a HNF1B en solo el 3% de los casos reconocidos, y la mayoría de los diagnosticados no tienen una causa genética. Entre las anomalías CAKUT asociadas con la trisomía 21 se incluyen las válvulas uretrales posteriores, la pielectasia y los megauréteres.29,30
Tabla 2 Un resumen de los genes implicados en el desarrollo de CAKUT.31,32,33,22,23,34,35,36
| Trastorno | Genes |
|---|---|
| Hipoplasia/Displasia renal | ACE, AGT, ATRX, CHD7, ESCO2, EYA1, SIX1, FRAS1, FREM2, GATA3, GLI3, GPC3, GRIP1, HNF1β, JAG1, KAL1, LRP4, MKS1–4, NIPBL, PAX2, PBX1, REN, SALL1, SIX5 |
| Agenesia renal | ATRX, BMP4, CBP/EP300, CHD7, ESCO2, FGF20, FRAS1, FREM2, GATA3, GRIP1, GLI3, HNF1β, ITGA8, JAG1, KAL1, KAL2, RET, SALL1, VANGL1 |
| Displasia quística | CBP/EP300, DHCR7, EVC, EVC2, FRAS1, FREM2, GPC-3, HNF1β, JAG1, KAL1, KAL2, MKS1–4, NS1, PEX, PAX2, WT1 |
| Sistema duplicado | NS1 |
| Riñón en herradura/Riñón ectópico | GLI3, NIPBL, VANGL1 |
| Hidronefrosis, UPJ | ACE, Adamts-1, AGT, ATRX, BMP4, CHD1L, CHD7, DHCR7, DSTYK, ESCO2, EYA, FRAS1, FREM2, GLI3, HSPG2, JAG1, Id2, NS1, PAX2, PEX, RET, SOX17, VANGL1 |
| Hidrouréter, Megauréter | CHD7, GLI3 |
| RVU | ATRX, DHCR7, EYA1, FOX1, GATA3, HOXA13, HPSE2, JAG1, KAL1, KAL2, MYH11, NIPBL, PAX2, SALL1, SIX1, SIX5 |
| VUP | BNC2, SALL1 |
Importancia clínica e implicaciones futuras del conocimiento genético
A medida que el campo de la evaluación genética continúa avanzando, es probable que veamos una capacidad creciente para identificar las causas genéticas de CAKUT y predecir la probabilidad de desarrollar estas afecciones. Esto podría conducir a un diagnóstico y tratamiento más tempranos, lo que podría mejorar los resultados para las personas afectadas.
Cabe señalar que las pruebas genéticas no siempre son necesarias para diagnosticar CAKUT. Por lo general, el diagnóstico definitivo se establece en función de una combinación de la presentación clínica, los antecedentes médicos y los estudios de imagen. Las pruebas genéticas para CAKUT suelen realizarse en un centro especializado bajo la supervisión de un genetista clínico, ya que la interpretación de estos resultados puede ser compleja y puede tener implicaciones para las decisiones reproductivas. Varios laboratorios (PreventionGenetics, Invitae, Blueprint Genetics, etc.) ofrecen pruebas genéticas para varios trastornos de CAKUT. Su panel de pruebas para CAKUT incluye pruebas genéticas de múltiples genes asociados con trastornos de CAKUT, incluidos HNF1B, NPHP1 y UPK1B. Las pruebas genéticas para estos trastornos pueden ayudar a confirmar un diagnóstico, proporcionar información sobre la gravedad del trastorno y ayudar en el asesoramiento genético y la planificación familiar. Cabe señalar que otras compañías y laboratorios de pruebas genéticas también podrían ofrecer CAKUT como un panel. Dado que CAKUT es un grupo de trastornos causados por diferentes mutaciones genéticas, el panel de pruebas genéticas para CAKUT en algunas compañías podría no incluir todas las causas genéticas de CAKUT, y distintos laboratorios pueden tener listas diferentes de genes cubiertos.
Una comprensión completa de los componentes genéticos de CAKUT es esencial para desarrollar estrategias precisas de pruebas genéticas. Además, esta comprensión ayudará a guiar la toma de decisiones clínicas en las pruebas genéticas. Se requieren estudios longitudinales que incluyan desenlaces clínicos significativos y función renal para investigar los efectos a largo plazo del diagnóstico y manejo precoces de CAKUT.
Conclusión y resumen
Las anomalías congénitas del riñón y del tracto urinario (CAKUT) son anomalías estructurales que afectan a los riñones y al tracto urinario. Se estima que hay alrededor de 4–60 casos por cada 10,000 nacimientos. El diagnóstico de CAKUT suele realizarse durante una ecografía prenatal o después del nacimiento en recién nacidos con signos y síntomas clínicos asociados. La anomalía más común es la obstrucción de la unión ureteropélvica, que afecta aproximadamente al 20% de las personas con la afección. El desarrollo de los riñones y del tracto urinario implica la formación y diferenciación de varias estructuras, incluidas las yemas ureterales y el metanefros, que deben conectarse adecuadamente para formar los riñones y el tracto urinario funcionales. Las alteraciones en este proceso pueden conducir a CAKUT. Existen diversas categorías de trastornos de CAKUT, incluidos los que afectan el número, la morfología y la posición de los riñones, y los que afectan el sistema colector. En el 10–25% de los casos, CAKUT es causada por trastornos genéticos. Se han identificado varios genes que están asociados con el desarrollo de diversas enfermedades renales. La investigación continúa para mejorar nuestra comprensión de las causas de CAKUT y desarrollar mejores estrategias de prevención y tratamiento. A pesar de sus limitaciones y dificultades actuales, el diagnóstico genético molecular puede brindar esperanza para mejorar el manejo clínico de los pacientes con CAKUT en el futuro.
Puntos clave
- Las anomalías congénitas del riñón y las vías urinarias (CAKUT) son un grupo de anomalías estructurales que afectan a los riñones y las vías urinarias, con una estimación de 4 a 60 casos por cada 10 000 nacimientos en la población general.
- El desarrollo de los riñones y las vías urinarias en el feto implica un proceso complejo que comienza en las primeras semanas del embarazo e incluye la formación y diferenciación de varias estructuras. Las alteraciones en este proceso pueden dar lugar a un espectro de anomalías congénitas.
- La prevalencia de CAKUT en neonatos prematuros fue del 2% en una cohorte amplia recientemente comunicada con 409 704 lactantes.
- La anomalía más frecuente es la obstrucción de la unión pieloureteral, que afecta aproximadamente al 20% de los individuos con la afección.
- En el 10-25% de los casos, CAKUT se atribuye a trastornos genéticos.
- La evaluación genética está avanzando, lo que aumenta la capacidad de identificar las causas genéticas de CAKUT y predecir la probabilidad de desarrollar estas afecciones.
- Las pruebas genéticas no siempre son necesarias para diagnosticar CAKUT; el diagnóstico suele basarse en una combinación de la presentación clínica, los antecedentes médicos y los estudios de imagen.
- Una comprensión completa de los componentes genéticos de CAKUT es esencial para desarrollar estrategias precisas de pruebas genéticas y orientar la toma de decisiones clínicas en pruebas genéticas.
- Se requieren estudios longitudinales para investigar los efectos a largo plazo del diagnóstico y manejo precoces de CAKUT.
Referencias
- Murugapoopathy V, Gupta IR. A Primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin J Am Soc Nephrol 2020; 15 (5): 723–731. DOI: 10.2215/cjn.12581019.
- Hays T, Thompson MV, Bateman DA, Sahni R, Tolia VN, Clark RH, et al.. The Prevalence and Clinical Significance of Congenital Anomalies of the Kidney and Urinary Tract in Preterm Infants. JAMA Netw Open 2022; 5 (9): e2231626. DOI: 10.1001/jamanetworkopen.2022.31626.
- Nicolaou N, Renkema KY, Bongers EMHF, Giles RH, Knoers NVAM. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol 2015; 11 (12): 720–731. DOI: 10.1038/nrneph.2015.140.
- Srivastava S, Molinari E, Raman S, Sayer JA. Many Genes–One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front Pediatr 2018; 5. DOI: 10.3389/fped.2017.00287.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. 8 (1): 25–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 2018; 128 (1): 4–15. DOI: 10.1172/jci95300.
- Gordon AC, Thomas DF, Arthur RJ, Irving HC. Multicystic dysplastic kidney: Is nephrectomy still appropriate? J Pediatr Surg 1988; 24 (9): 951. DOI: 10.1016/s0022-3468(89)80673-6.
- Schreuder MF, Westland R, Wijk JAE van. Unilateral multicystic dysplastic kidney: a meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol Dial Transplant 2009; 24 (6): 1810–1818. DOI: 10.1093/ndt/gfn777.
- Kopač M, Kordič R. Associated Anomalies and Complications of Multicystic Dysplastic Kidney. Pediatr Rep 2022; 14 (3): 375–379. DOI: 10.3390/pediatric14030044.
- Fletcher J, Hu M, Berman Y, Collins F, Grigg J, McIver M, et al.. Multicystic Dysplastic Kidney and Variable Phenotype in a Family with a Novel Deletion Mutation ofPAX2. J Am Soc Nephrol 2005; 16 (9): 2754–2761. DOI: 10.1681/asn.2005030239.
- Chang Y-M, Chen C-C, Lee N-C, Sung J-M, Chou Y-Y, Chiou Y-Y. PAX2 Mutation-Related Renal Hypodysplasia: Review of the Literature and Three Case Reports. Front Pediatr 2022; 9 (765929). DOI: 10.3389/fped.2021.765929.
- Bienstock JL, Birsner ML, Coleman F, Hueppchen NA. Successful In Utero Intervention for Bilateral Renal Agenesis. Obstet Gynecol 2014; 124 (2): 413–415. DOI: 10.1097/aog.0000000000000339.
- Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, et al.. Faculty Opinions recommendation of Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2012; 1 (2): 96–200. DOI: 10.3410/f.13934957.15391059.
- Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal Aplasia in Humans Is Associated with RET Mutations. Am J Hum Genet 2008; 82 (2): 344–351. DOI: 10.1016/j.ajhg.2007.10.008.
- Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al.. Faculty Opinions recommendation of Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2006; 7 (10): 864–870. DOI: 10.3410/f.1047767.508721.
- LEBOWITZ ROBERTL, GRISCOM NTHORNE. Neonatal Hydronephrosis. Radiol Clin North Am 1977; 15 (1): 49–59. DOI: 10.1016/s0033-8389(22)02539-8.
- AC SJ, DM M, oes S, AC S. Congenital Anomalies of the Kidney and Urinary Tract: An Overview. Congenital Anomalies of the Kidney and Urinary Tract 2014; 02 (4): 1–13. DOI: 10.1007/978-3-319-29219-9_1.
- Avanoglu A, Tiryaki S. Embryology and Morphological (Mal)Development of UPJ. Front Pediatr 2020; 8: 137. DOI: 10.3389/fped.2020.00137.
- Aoki Y, Mori S, Kitajima K, Yokoyama O, Kanamaru H, Okada K, et al.. Id2 haploinsufficiency in mice leads to congenital hydronephrosis resembling that in humans. Genes Cells 2004; 9 (12): 1287–1296. DOI: 10.1111/j.1365-2443.2004.00805.x.
- Williams G, Fletcher JT, Alexander SI, Craig JC. Vesicoureteral reflux. Journal of the American Society of Nephrology 2008; 19 (5): 847–862. DOI: 10.1681/asn.2007020245.
- Darlow JM, Darlay R, Dobson MG, Stewart A, Charoen P, Southgate J, et al.. Genome-wide linkage and association study implicates the 10q26 region as a major genetic contributor to primary nonsyndromic vesicoureteric reflux. Sci Rep 2017; 7 (1): 59. DOI: 10.1038/s41598-017-15062-9.
- Woolf AS, Lopes FM, Ranjzad P, Roberts NA. Congenital Disorders of the Human Urinary Tract: Recent Insights From Genetic and Molecular Studies. Front Pediatr 2019; 7 (136). DOI: 10.3389/fped.2019.00136.
- Wu C-HW, Mann N, Nakayama M, Connaughton DM, Dai R, Kolvenbach CM, et al.. Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT). Genet Med 2020; 22 (10): 1673–1681. DOI: 10.1038/s41436-020-0844-z.
- Verbitsky M, Krithivasan P, Batourina E, Khan A, Graham SE, Marasà M, et al.. Review of: "Genome-wide association study followed by trans-ancestry meta-analysis identify 17 new risk loci for schizophrenia". J Am Soc Nephrol 2021; 32 (4): 805–820. DOI: 10.32388/jqb9pr.
- Thakkar D, Deshpande AV, Kennedy SE. Epidemiology and demography of recently diagnosed cases of posterior urethral valves. Pediatr Res 2014; 76 (6): 560–563. DOI: 10.1038/pr.2014.134.
- Warshaw BL, Edelbrock HH, Ettenger RB, Malekzadeh MH, Pennisi AJ, Uittenbogaart CH, et al.. Renal transplantation in children with obstructive uropathy. J Pediatr Surg 1980; 15 (6): 986. DOI: 10.1016/s0022-3468(80)80355-1.
- Roth KS, Carter WH, Chan JCM. Obstructive Nephropathy in Children: Long-Term Progression After Relief of Posterior Urethral Valve. Pediatrics 2001; 107 (5): 1004–1010. DOI: 10.1542/peds.107.5.1004.
- Hennus PML, Kort LMO de, Bosch JLH, Jong TPVM de, Heijden GJMG van der. A Systematic Review on the Accuracy of Diagnostic Procedures for Infravesical Obstruction in Boys. PLoS One 2014; 9 (2): e85474. DOI: 10.1371/journal.pone.0085474.
- Rodriguez MM. Congenital anomalies of the kidney and urinary tract (CAKUT). Lijec Vjesn 2014; 144 (Supp 1). DOI: 10.26800/lv-144-supl1-26.
- Postolache L, Parsa A, Simoni P, Boitsios G, Ismaili K, Schurmans T, et al.. Widespread kidney anomalies in children with Down syndrome. Pediatr Nephrol 2022; 37 (10): 2361–2368. DOI: 10.1007/s00467-022-05455-y.
- Uy N, Reidy K. Developmental Genetics and Congenital Anomalies of the Kidney and Urinary Tract. J Pediatr Genet 2016; 05 (01): 051–060. DOI: 10.1055/s-0035-1558423.
- Kolvenbach CM, Dworschak GC, Frese S, Japp AS, Schuster P, Wenzlitschke N, et al.. Expanding congenital abnormalities of the kidney and urinary tract (CAKUT) genetics: basonuclin 2 (BNC2) and lower urinary tract obstruction. Ann Transl Med 2019; 7 (S6): S226–s226. DOI: 10.21037/atm.2019.08.73.
- Heidet L, Morinière V, Henry C, De Tomasi L, Reilly ML, Humbert C, et al.. Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 2017; 28 (10): 2901–2914. DOI: 10.1681/asn.2017010043.
- Talati AN, Webster CM, Vora NL. Prenatal genetic considerations of congenital anomalies of the kidney and urinary tract (CAKUT). Prenat Diagn 2019; 39 (9): 679–692. DOI: 10.1002/pd.5536.
- Puri P, Gosemann J-H, Darlow J, Barton DE. Genetics of vesicoureteral reflux. Nat Rev Urol 2011; 8 (10): 539–552. DOI: 10.1038/nrurol.2011.113.
- Chen F. Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol 2009; 24 (9): 1621–1632. DOI: 10.1007/s00467-008-1072-y.
Última actualización: 2025-09-21 13:35