
22: Cloacal Exstrophy
This chapter will take approximately 30 minutes to read.
Introduction
Cloacal exstrophy (CE) commonly referred to as the omphalocele, exstrophy of the bladder, imperforate anus, and spinal abnormalities (OEIS) complex, is the least common manifestation of the exstrophy-epispadias complex (EEC), however, it represents the greatest divergence from typical anatomy and development. As a multisystem defect, it requires a dedicated multidisciplinary approach to achieve optimal outcomes for patients and their families. This chapter provides a review of cloacal exstrophy through a modern lens alongside future directions in understanding this burdensome condition.
Embryology
The prevailing theory behind the EEC is the failure of mesodermal ingrowth to reinforce the cloacal membrane.1 The cloacal membrane is a bilaminar layer of ectodermal and endodermal tissue located at the caudal aspect of germinal disk of the developing infraumbilical abdominal wall. In normal development, during the 4th and 5th weeks of gestation, mesenchymal ingrowth between these layers forms the lower abdominal muscles and bony pelvis. Continued caudal descent of this urorectal septum results in fusion with the cloacal membrane and, ultimately, separation of the cloaca into the bladder anteriorly and rectum posteriorly (Figure 1).2 Perforation of the cloacal membrane typically occurs after fusion with the urorectal septum at approximately the 6th week of development, forming separate urogenital and anal orifices. The paired genital tubercles that give rise to the phallus, then migrate medially and fuse in the midline.3
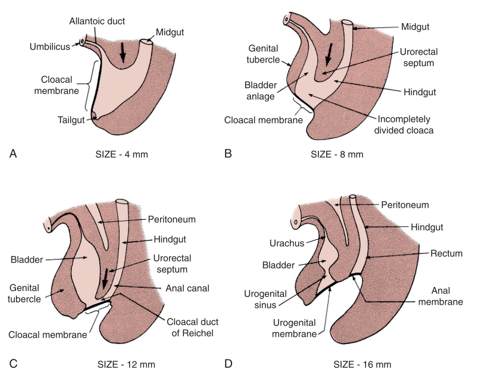
Figure 1 Development of cloacal membrane, urogenital sinus, and anal openings. Mesenchymal ingrowth with caudal descent of the urorectal septum and perforation of the cloacal membrane resulting in the urogenital and anal cavities.
The lack of mesenchymal reinforcement, and resultant overdevelopment of the cloacal membrane, risks premature rupture of the cloacal membrane. The timing, and degree at which premature rupture of this membrane occurs, results in the spectrum of conditions seen in EEC; CE is the earliest forming defect.
Epidemiology
The incidence of CE is approximately 1 in 200,000-400,000 live births4,5 Historically in CE, there is a male-to-female predilection of 2:1, though some series place this closer to 1:1.6,7,8 With improved imaging modalities and prenatal diagnosis, however, the number of CE live births has markedly decreased as pregnancies are electively terminated in 23% of cases and up to 50% of pregnancies spontaneously abort.9
Most cases appear sporadic, however, genetic analyses have identified possible causative factors. Thauvin-Robinet et al described an unbalanced translocation between the long arm of chromosome 9 and the Y chromosome resulting in a 9q34.1-qter deletion as a possible cause, while mutations in a group of homeobox genes including HLXB9 and the HOX family, impacting mesodermal development, were implicated by others.10
In addition to family history, other CE-specific risk factors have been implicated. In a clinical and molecular analysis of 232 families, advanced parental age emerged as a possible risk factor with an average maternal age of 34 years and average paternal age of 32 years (compared to 26 and 27 years based on 2010 census data, respectively). Interestingly, 49% of exstrophies were born from first pregnancies, which may also reinforce advancing parental age.10 Hormonal influences may also play a role as Wood and colleagues identified a 10-fold increase in exstrophy births to mothers who received large doses of progesterone early in the first trimester.11 In this same study, associations with assisted reproductive techniques were also reported as children conceived with in vitro fertilization (IVF) experienced a 7.5-fold increase in exstrophy.
Currently, the environmental factor most closely linked to cloacal exstrophy is maternal tobacco exposure, specifically periconception maternal exposure to smoking and maternal smoking during the first trimester.12 Taken together two critical points in counseling arise: 1) appropriate counseling regarding optimizing the periconception and first trimester periods – during the time of organogenesis and 2) an understanding of shifting societal views towards family planning (with first-time parents advancing in age) and the potential need for assisted-reproductive technologies.
Pathogenesis and Associated Anomalies
The complexity in determining the multifactorial causes underlyingCE is demonstrated in the range of defects observed in this phenotype (Table 1). As CE is the result of early developmental events, it presents with more anomalies to the severest degree and represents the most challenging end of the exstrophy-epispadias spectrum. The classic phenotypic expression of CE involves a strip of exstrophied cecum flanked by two exstrophied bladder halves which are often accompanied by a prolapsed ileal segment lending to the “elephant trunk” deformity (Figure 2). Additionally, the hindgut is shortened and often blind-ending, resulting in an imperforate anus. As the genital tubercles fail to fuse in the midline, two phallic halves are seen on either side of a widened pubic diastasis (Figure 3). The abdominal wall defect is generally accompanied by an omphalocele of varying size.2 Due to the multisystemic nature of this defect, each anomaly is described in more detail separately.
Table 1 Cloacal Exstrophy and its associated anomalies.
| Gastrointestinal | Genitourinary | Central Nervous System | Musculoskeletal |
|---|---|---|---|
| Omphalocele | Unilateral renal agenesis | Tethered cord | Vertebral (absent or hemi defects) |
| Imperforate anus, anal atresia or stenosis | Pelvic kidney | Myelomeningocele | Club foot |
| Short gut syndrome | Ureteral duplication | Hip subluxation | |
| Intestinal malrotation | Hydronephrosis | Paucity of pelvic floor musculature anteriorly | |
| Intestinal duplication | Bilateral cryptorchidism | ||
| Inguinal hernias | |||
| Uterine duplication | |||
| Vaginal duplication |
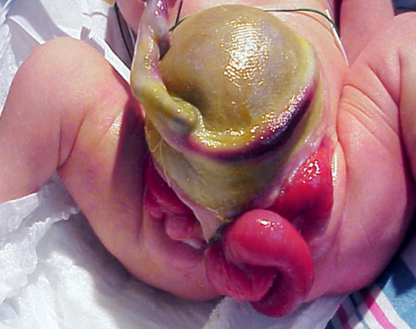
Figure 2 Male with classic presentation of cloacal exstrophy with prolapsed ileal segment creating the “elephant trunk” deformity.
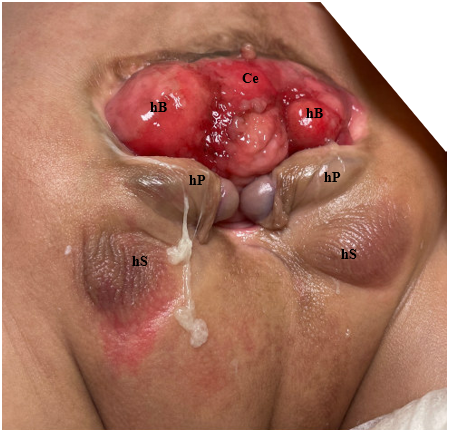
Figure 3 Photograph of male with cloacal exstrophy. Note, a colostomy has already been performed. Ce = cecum; hB = hemibladder; hP = hemiphallus; hS = hemiscrotum.
Musculoskeletal
Bony Pelvis
Anomalies of the bony pelvis and the associated pubic diastasis are the hallmark of exstrophy. In 1995, Sponseller et al utilized three-dimensional (3D) computed tomography (CT) of the bony pelvis in exstrophy patients and elucidated abnormalities that modernized our understanding of the characteristic widening of the pubic symphysis. Most notably, they identified two broad categories of abnormalities, deemed rotational and dimensional anomalies (Table 2).
Table 2 Anomalies of the Bony Pelvis.
| Rotational | Dimensional |
|---|---|
| External rotation of the posterior pelvis/iliac wings | Increased pubic diastasis |
| External rotation of the anterior pelvic segment | Shortened anterior pubic segment (30%) |
| Coronal rotation of the sacroiliac joint | Increased intertriradiate cartilage distance |
| Acetabular retroversion | |
| Convergence of iliac wings | |
| Femoral retroversion |
Compared to age-matched controls, Sponseller et al found that, in classic bladder exstrophy (CBE), the posterior pelvis is externally rotated an average of 12-degrees on each side, with the anterior pelvis externally rotated an average of 18-degrees. Additionally, the pubic rami are 30% shorter, which, combined with a retroverted acetabulum, leads to an average diastasis of 4.8 cm in CBE (Figure 4).13 In CE patients, these defects are even more exaggerated, with an overall 43% decrease in length of bone in the entire pelvis, with an average pubic diastasis greater than 6 cm and an increased likelihood of asymmetry between the right and left sides of the pelvis. For clarity, mild diastasis is less than 4 cm, moderate diastasis is between 4 and 6 cm and extreme diastasis is anything > 6 cm.
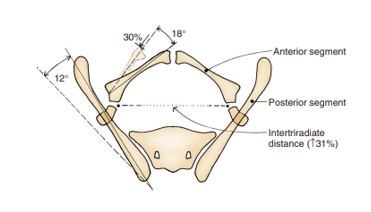
Figure 4 Pelvic bone anomalies noted in classic bladder exstrophy. The posterior bone segment is externally rotated 12˚ on each side, but the length is unchanged. The anterior segment is externally rotated 18˚ on each side and shorted by 30%. The distance between the triradiate cartilage is increased by 31%. Note, anomalies are more severe in cloacal exstrophy and asymmetry is more common. Used with permission Brady Urological Institute.
Pelvic Floor Defects
The rotational and dimensional anomalies of the bony pelvis directly impact the maldevelopment of the pelvic floor. 3D-CT imaging of children with exstrophy revealed that the levator ani muscles are positioned more posteriorly (68% posteriorly/32% anteriorly) compared to age-matched controls (52% posteriorly/48% anteriorly) (Figure 5).14 In addition to being more posteriorly oriented, the levator ani is also less concave. Another consequence of the bony pelvis deformities manifested in the pelvic floor is the more flattened puborectalis compared to its usual conical shape. The paucity of anterior pelvic floor musculature and the lack of a conical shape drive the surgical technique in reconstructing the bony pelvis and pelvic floor, which is discussed separately.
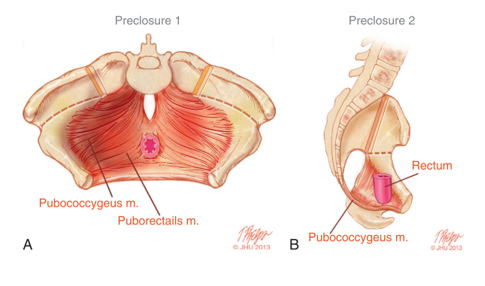
Figure 5 A) Pelvic floor musculature prior to exstrophy closure with osteotomy incisions marked. B) Lateral view of pelvic floor anatomy showing posterior displacement of pelvic floor muscles behind rectum.
Lower Limb and Gait Abnormalities
Abnormalities of the lower extremities are consistently encountered in CE. As a result of the increased pubic diastasis, distance between the hips, and external rotation of the acetabula, children may compensate with outward rotation of the lower limbs and a waddling gait. Additional deformities include tibial torsion, equinovarus deformities, and calcaneus deformities.15 Despite these limb deformities, range of motion in these patients is usually not affected and as-needed corrective braces help correct the gait over time. By 2 years of age, most children are ambulatory with minimal use of assistive devices and few require wheelchair assistance in childhood.
Gastrointestinal Anomalies
Gastrointestinal anomalies occur to some degree in nearly all patients and add significantly to the morbidity and the degree of difficulty in managing these patients, both in the perioperative setting with respect to nutrition and in the long-term. Omphalocele is characteristic of this condition and found in 88–100% of cases.16,17,18,19 Omphaloceles may vary greatly in size and their contents, containing small bowel, liver, or both. Immediate closure of the omphalocele defect or housing it within a protective silo in the newborn period is typically performed to prevent subsequent rupture while providing a hospitable environment. In a series by Davidoff et al which included 26 patients with CE, all presented with an omphalocele. Of these patients, 19 (73%) were closed primarily with the remainder requiring staged reduction with a temporary prosthetic silo.16
Short bowel syndrome, present in 25% of cases, is a pervasive cause of increased nutritional morbidity and may occur despite normal bowel length.4 A potential inherent absorptive abnormality of the intestine further underscores optimal perioperative nutrition in these patients and the need to preserve as much large bowel length as possible during reconstruction. If not utilized for stool, the hindgut remnant may be incorporated during reconstruction of the urogenital tract.17 While numerous pathways exist for initial management of the intestinal tract, work by Sawaya et al led to a paradigm shift in 2009 with initial tubularization of the cecal plate with end colostomy placement as this drastically reduced the occurrence of short gut syndrome and enabled future pull-through procedures in eligible candidates.20
Other associated anomalies of the gastrointestinal tract are seen in 46% of cases and include duplication anomalies, gastroschisis, malrotation, duodenal atresia, and exstrophied colonic segments.16,17,21 Imperforate anus is also widely encountered, with one series noting its presence in 36 of 37 patients.17
Other Urologic Anomalies
Anomalies of the upper urinary tract are common, occurring in 41–66% of patients.17,18 Particular attention to identifying and understanding the patient’s upper tract anatomy is vital as unilateral renal agenesis, pelvic kidney or hydronephrosis can be seen in up to 48% of cases, with multiple concomitant renal anomalies in 16% of patients.(missing reference) Horseshoe kidneys, fusion anomalies, and ureteral abnormalities including megaureter and duplication are reported less frequently. Of note, ureteral anomalies can vary widely with possible ectopic ureteral insertion to the vas deferens in the male and into the uterus, vagina, or fallopian tubes in females.19
Central Nervous System and Vertebral Anomalies
Spinal dysraphism, including tethered cord, myelomeningocele, or lipomyelomeningocele, is present in some form in 64-100% of patients, with 80% found in the lumbar region.2,18 Vertebral anomalies other than dysraphism include hemi-vertebrae and associated scoliosis, which is seen in 40% of patients, though the scoliosis does not progress in most cases.15 Due to this high incidence of spinal and vertebral defects, MRI evaluation of a newborn with CE is warranted with neurosurgical involvement as needed.
Neurological impairment may ultimately affect bladder function, urinary continence, and lower extremity movement. The degree to which each may be affected is just as variable as the spinal and vertebral anomalies. Further, microdissection of an infant pelvis with CE in the post-mortem setting revealed abnormal origin and vascular supply of the autonomic nerves.22 This neurological unpredictability adds to the challenge in helping these patients achieve continence. Abdominal Wall Defects
The premature rupture of the cloacal membrane may result in a large defect with complete absence of the lower abdominal wall. While the presentation of this defect is readily apparent, subtleties require particular attention. As a result of the widened pubic diastasis and changes to the typical oblique orientation of the inguinal canal, inguinal hernias are noted in up to 50% of patients with CE and should be repaired.23
Anomalies Affecting Reproductive Organs and Genitalia
In the male CE patient, complete asymmetric separation of the phallus into two halves with concordant separation of the scrotal halves is typical. The testes may be located within their respective hemiscrota; however, they are often undescended and associated with inguinal hernias, requiring surgical correction. In females, the clitoral bodies are similarly widely separated.
Müllerian anomalies are also common in CE, with uterine duplication seen in 95% of cases.19 Partial duplication predominates with bicornate uterus being the most common subtype. Vaginal anomalies range from agenesis, seen in 25-50% of patients, to vaginal duplication, which occurs in up to 65% of cases.21 In instances of uterine or fallopian tube duplication, preservation should be prioritized for possible incorporation into lower urinary tract reconstruction.
Cardiovascular and Pulmonary Anomalies
Rarely, CE presents with immediately life-threatening cardiovascular or pulmonary defects. Reports, though rare, include a plethora of major anomalies including aortic duplication and caval duplication.23 Others have described Tetralogy of Fallot, pulmonary atresia, pulmonary artery stenosis, and patent ductus arteriosus.24
Given the associated impact of this birth defect on the development of nearly every organ system, care for these vulnerable patients should be provided at a center experienced in the surgical techniques with cross-specialty perioperative and postoperative management.
Evaluation and Diagnosis
Making the Diagnosis
Considering the range of defects associated with CE, prenatal diagnosis has historically proved challenging, if not elusive. Accurately diagnosing CE and distinguishing it from CBE is paramount in properly counseling families, especially considering that children with CE tend to face more health-related issues in their lifetime. Prenatal diagnosis allows for planning that may optimize postnatal medical and surgical management. The first prenatal diagnosis of CE in 1985 relied on three key fetal ultrasound (fUS) findings: 1. A large midline infraumbilical anterior abdominal defect, 2. lumbosacral myelomingocele, and 3. failure to visualize the urinary bladder.25 Austin and colleagues further refined this list by delineating diagnostic criteria as either major or minor findings. Seen in >50% of cases constituted a major criterion and included: non-visualization of the bladder (91%), a large midline infraumbilical anterior wall defect or cystic anterior wall structure (82%), omphalocele (77%), and myelomeningocele (68%). Minor criteria were those seen in <50% of cases and included: lower extremity defects (23%), renal anomalies (23%), ascites (41%), widened pubic arches (18%), narrow thorax (9%), hydrocephalus (9%), and a single umbilical artery (9%).26 Despite vast imaging improvements in fUS since it was first introduced as a diagnostic tool, only an estimated 15% of patients have been diagnosed prenatally on fUS alone as findings may be incompletely identified as isolated omphalocele, CBE, or other midline defects.
The advent of fetal magnetic resonance imaging (fMRI) in prenatal diagnosis has added a valuable adjunct in evaluating CE. Compared to fUS, fMRI provides superior anatomical visualization when a bladder is not identified and may also aid in assessing the presence or absence of an omphalocele, associated spinal defects, and gender, when not readily identified on US.27,28
Recently, Weiss et al identified key anatomic findings on fUS and fMRI to assess their respective validity in prenatally diagnosing CBE and CE. Between 2001 and 2018 they identified 21 patients who had prenatal imaging. CBE was the postnatal diagnosis in 14 and CE in the remainder. 15 of 21 patients had both fUS and fMRI available for review and the median gestational age for evaluation by prenatal imaging was 25 weeks. Of the 16 fUS with initial interpretations available, the original prenatal diagnosis was correct in 12 cases, yielding a 69% sensitivity of fUS. All 4 cases of incorrect prenatal diagnoses of CE were later determined to be CBE. Of the 18 fMRIs included in analysis, 16 of 18 diagnoses aligned (83% sensitivity) and the two incorrect prenatal CE diagnoses were reclassified as CBE. These misdiagnoses were attributed to a large protruding bladder plate with bowel loops posteriorly imitating an omphalocele containing bowel. They concluded that identification of the point of umbilical cord insertion on both fUS and fMRI was prudent to differentiate between CBE and CE given the abdominal wall defects.29
Variants in cloacal exstrophy provide additional diagnostic challenges. Though exceedingly rare, a skin covered variant is also possible, as described in 5 of 6 patients in one small case series.30
Evaluation and Immediate Postnatal Management
After birth, it is essential to ensure that the patient is medically stable. A thorough physical examination is required to confirm the various anatomic anomalies that may be present (Table 1). Elucidating the associated birth defects and their severity aids in medical and surgical management. As with CBE patients, the exstrophied bowel and bladder segments are kept moist protected by plastic dressing.31 A complete imaging series, including plain films, US and MRI, helps determine the severity of associated defects and facilitates early involvement of consulting services such as pediatric orthopedic surgery, pediatric neurosurgery, and pediatric general surgery. Rounding out the multidisciplinary team with input and support from social work and nutrition services is key to caring for the infant with CE and their family.
Gender Assignment
Once the infant is medically stable and the complete extent of the condition is understood, an honest and thoughtful discussion regarding gender assignment is necessary. In many instances, this may require consultation from pediatric endocrinology, child psychology, or pediatric psychiatry. All gender assignment decisions should only be made after karyotyping, thorough discussion, and appropriate parental counseling.
Given the typical phenotypic expression with wide separation of the penile bodies and scrotum and diminutive corporal size in boys with CE, initial reports advocated universal gender reassignment of 46, XY boys to functional females.32 This drove early principles in management, including bilateral orchiectomy along with phallic reconstruction as a functional clitoris and vaginoplasty, either early or delayed.
The long-term effects of this practice are currently the topic of intense debate. Patients living longer with this condition offer a glimpse into the psychosocial implications of this practice and potential role of genotype and intrauterine hormonal milieu. In one cohort of 29 males with CE who underwent female gender reassignment, all 29 patients demonstrated a predominant male shift in psychosexual development despite not experiencing any pubertal hormonal surges.33 Other series, however, have demonstrated no differences in behavior or psychosocial issues, although there was one reported case of masculinization in a 46XY gender converted patient due to an ectopic testis.34,35 Current attitudes favor assigning gender that is concordant with karyotype, if possible. This was supported by a survey of pediatric urologists in which two-thirds favored gender-congruent reconstruction.36
Surgical Management and Outcomes
Initial Management
Prompt omphalocele closure in the neonatal period is recommended to
safeguard against untimely rupture; however, this is only performed after neurosurgical concerns are addressed. At the time of initial omphalocele closure, 1 of 3 pathways is typically employed for intestinal diversion and hindgut management, including ileostomy creation with hindgut resection, ileostomy placement with hindgut mucous fistula, or cecal tubularization with end-colostomy creation.
Historically, initial intestinal diversion relied on ileostomy with hindgut resection, however this created several unintended consequences. Firstly, this induced short gut syndrome universally and made later reconstruction of the gastrointestinal tract less likely. Secondly, high-output ileostomies predisposed children to increased hospitalization with recurrent dehydration and electrolyte derangements.18,20 The induced acidosis secondary to high ileostomy output adversely impacts calcium homeostasis and directly modulates the growth hormone-IFG-1 axis, blunting the release of growth hormone. Long-term, this is associated with a growth-related morbidity in CE patients.37
Due to this, there has been a paradigm shift in initial intestinal diversion and hindgut management. In their series of 77 patients, Sawaya et al found hindgut length varied widely from 2 to >20 cm with over half in the 6 to 15 cm range. Interestingly, patients with genotype XY were more likely to have a hindgut length less than 10 cm compared to their XX counterparts. In this series, only 10 patients underwent hindgut resection and only for questionable hindgut viability or extremely short length. As a result, the authors advocated cecal tubularization with end colostomy creation to eliminate short gut syndrome and facilitate intestinal pull-through procedures.20 Typically, gastrointestinal reconstruction is performed 1 to 2 years after initial fecal diversion, however, if this reconstruction is combined with bladder closure, approximation of the pubis is paramount for successful bladder, abdominal wall, and bowel reconstruction. Typically, success of these reconstructions rely on pelvic reconstruction with osteotomies.17
In their same series, Sawaya et al performed eight intestinal pull-through procedures—7 by the age of 5 years, ranging from 2 to 12 years of age—and all patients had preserved hindguts of at least 10 cm in length.20 Other factors to consider when deciding whether to perform intestinal pull-through include ability of patient to make solid stool and evidence of anal sphincteric musculature responsive to stimulation when examined under anesthesia. In children who are not candidates for intestinal pull-through, a permanent fecal stoma is the most durable option for long-term management. Of note, if the hindgut remnant is not incorporated in the fecal stream, it should be preserved for later bladder augmentation or vaginal reconstruction.17
If it is determined at the initial stage of omphalocele closure that dual bladder and abdominal wall closure cannot be achieved safely and successfully, then the bladder halves should be approximated in the midline, converting the defect to a CBE.17 This allows abdominal distention and enlargement of the bladder plate for subsequent delayed closure. Finally, the hindgut is left as a mucous fistula at this time if not incorporated in the initial bowel reconstruction.
Urinary Reconstruction
Modern Staged Repair
Urinary reconstruction of CE parallels that of CBE. Approximation of the hemibladders posteriorly converts the defect from CE to CBE and involves dissection of the lateral aspects of the bladder halves from the abdominal wall, and closure in the midline.38 Restoring anatomy via placement of the bladder and posterior urethra deep into the pelvis remains a crucial factor in successful surgical reconstruction. Approximation of the widened pelvis aids in this goal, permitting abdominal wall and urinary tract reconstruction, however, this usually requires pelvic osteotomies and fixation, as will be described later in this chapter.
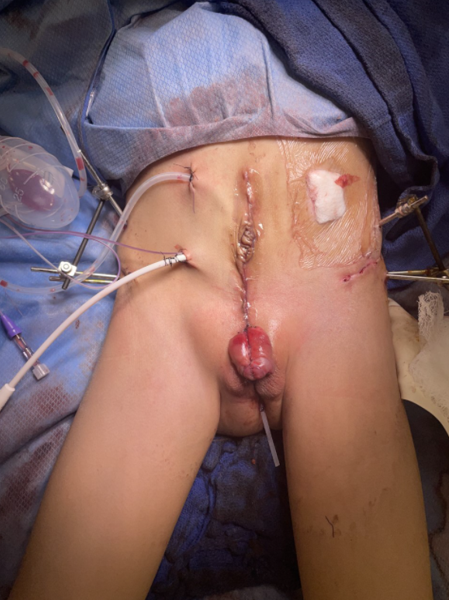
Figure 6 Modern staged repair of exstrophy. Postoperative photograph of patient seen in preoperative photograph in Figure 3. Note osteotomy pin placement prior to application of external fixation device.
The large abdominal wall defect found in CE presents a significant challenge in tension-free abdominal wall reconstruction (Figure 7). Failure to consider tension at time of closure may impact rates of successful closure and fistula formation. Bioprosthetic materials such as Alloderm (Allergan, Branchburg, NJ) have shown great promise in bridging the gap in lack of abdominal wall tissue while providing a tension-free closure.39 Additionally, Alloderm has been used as an adjunct to decrease penopubic fistulization through coverage of the interpubic stitch at time of urinary tract reconstruction.40
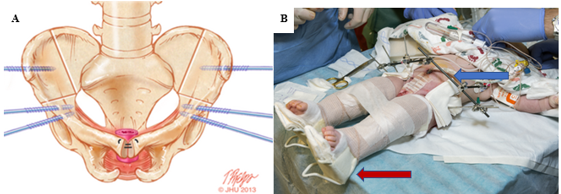
Figure 7 A, combined bilateral anterior innominate and vertical iliac osteotomy locations marked in bone with ideal placement of pin insertions. B, modified Buck’s traction (red arrow) and external fixation (blue arrow) at right.
To ensure congruence with the assigned gender, reconstruction of the external genitalia is performed as soon as possible in the immediate postnatal period. In genotypic males, this will ensure they are raised congruent to their phenotype. While the long-term psychosocial and psychosexual effects of children who undergo gender reassignment is currently the subject of great interest, as discussed earlier, histological studies of testes in male subjects who underwent gender reassignment confirm normal histology, even despite cryptorchidism.41 Given the often demure and asymmetric penile tissue present, phallic reconstruction remains challenging with mixed results.
Phalloplasty has emerged as a successful reconstruction option for penile replacement, though this is typically delayed until late adolescence (Figure 8).42 However, in instances where the paucity of tissue requires male-to-female reassignment, initial genital reconstruction should adjoin the phallic halves in the midline to form a clitoris. When phallic tissue is adequate, however, epispadias repair is performed at 1 year of age, often using the well-described Cantwell-Ransley Repair.43 In genetic females, vaginal reconstruction is performed early, and in gender reassigned males, delayed reconstruction of a neovagina is appropriate with long-term dilation of the neovagina required.44 Due to the shorter urethra in genetic females, repair of isolated epispadias is typically combined with Young-Dees-Leadbetter bladder neck reconstruction (BNR), monsplasty, and clitoroplasty.
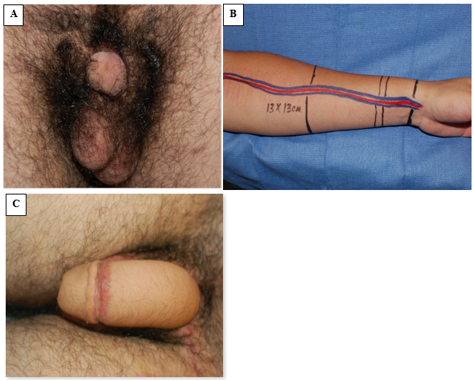
Figure 8 Radial forearm free flap phalloplasty. A, preoperative photograph with demure penile tissue. B, intraoperative preparation of forearm flap tissue and associated pedicled flap vasculature. C, postoperative photograph demonstrating outcome of phalloplasty.
The Importance of Osteotomy
Osteotomy has emerged as a critical factor in successful closure in most children with CBE is indicated in all children with CE at time of bladder closure.17,44 Osteotomy corrects the wide pubic diastasis and allows for orthotopic positioning of the bladder and posterior urethra as deep as possible within the pelvis while also facilitating bladder and abdominal wall reconstruction with minimal tension. This reduced tension correlates with decreased rates of dehiscence and postoperative ventral hernias. Additionally, osteotomy has been found to reduce the likelihood of significant complications from 89% in patients closed without osteotomy to 17% in children closed with osteotomy.45 This was confirmed in a large series of 80 CE patients that reported 91% successful exstrophy repairs with osteotomy performed at time of closure.46 While osteotomy has aided in successful closure, it has not been found to impact eventual continence in patients with CE.
Combined bilateral anterior innominate and vertical iliac osteotomies are the preferred method at our institution and is coordinated with bladder closure in an approach coined the Dual-Staged Pathway (DSP).47 This approach to osteotomy in the supine position provides the added benefit of not needing to reposition the patient prior to bladder and abdominal wall closure (Figure 7). As a posterior approach is avoided, the risks of damaging any spinal repair are also minimized. Additionally, pelvic osteotomies allow gradual pelvic reduction with external fixation 2 to 3 weeks prior to bladder and abdominal wall closure. This is often utilized in instances of extremely wide pubic diastasis (> 10 cm) and was successfully used in one patient with a pubic diastasis of 16 cm.47 At our institution, external fixation and modified Buck’s traction are maintained for 6 to 8 weeks to ensure adequate healing. In our experience, this immobilization technique provides exceptional results with a 3.8% failure rate in primary closures, compared to 65.7% for immobilization with a spica cast.48
Single-Stage Reconstruction
Another approach to repair in the patient with CE is the single-step repair, or complete primary repair of exstrophy (CPRE), popularized by Grady and Mitchell.49 Unlike the staged approach of abdominal and bladder reconstruction, followed by epispadias repair and a continence procedure, the single-staged repair combines aspects from each stage into a single repair. The procedure is similar to the one described for CBE, however it considers timing of closure with respect to the omphalocele, emphasizing delay in closure for a large omphalocele or other extenuating medical issues. Others have concluded that while single-staged repair is applicable to a select group of patients, other factors that may preclude its use are a small bladder plate, pubic diastasis >6 cm, or other medical conditions such as spinal dysraphism, which, even in the absence of exstrophy, may require multiple surgical procedures .50 In a limited series of 6 patients, Lee et al reported successful closure with complete primary repair. They did note, however, the development of a midline bladder septum with associated hydronephrosis in two children. Additionally, one child voided naturally and another underwent augmentation cystoplasty (AC).51
Proponents of this technique advocate that it may decrease cost, the morbidity and trauma associated with multiple operations, and even stimulate early bladder growth. Epispadias repair in this instance is performed via penile disassembly wherein the urethral plate is dissected entirely from the corporal bodies. This allows urethral closure and positions the bladder neck posteriorly within the pelvis.52 CPRE advocates attribute the decreased number of operations to achieve continence to increased bladder outlet resistance at an early age. However, many patients still require BNR and, notably, only 20-56% of CPRE patients who remain incontinent achieve continence following additional BNR.53,54
Complications
Both single and staged repairs are acceptable reconstruction techniques in CE. Though each approach has its merits, reported outcomes place the failure rate for a single-stage reconstruction at 48% compared to 15% for a staged repair. Wound dehiscence, bladder prolapse, bladder outlet obstruction, and formation of a vesicocutaneous fistula should all be considered failed closures and have been reported in both single and staged techniques.
With either surgical approach, successful primary bladder closure is the primary goal. A failed closure is devastating for the patient and family and the ramifications are significant. Patients that experience a failed primary closure in CE are subject to more operations and general anesthetic exposure. Though a failed bladder closure in CBE negatively impacts ultimate urinary continence, this impact on continence specifically is less pronounced in the patients with CE as most of these patients require additional continence procedures regardless of primary outcome.55,56 The ramifications extend beyond the additional operations to include financial morbidity. Goldstein et al performed a cost analysis in CE patients and found that with a successful primary repair of CE it costs approximately $196,000 to reach continence, while the cost to achieve continence rises to $407,000 after a failed primary bladder closure.57
One large series assessing predictors of successful primary closure in exstrophy reported that a staged CE closure, along with the use of pelvic osteotomy, were significant independent predictors of a successful closure. Additionally, after bladder closure, postoperative immobilization was crucial to success.58 For this reason, we recommend the use of external fixation combined with traction as is the norm at our institution. Other methods, such as spica casts may not provide adequate immobilization and may, therefore, lead to increased failure of pelvic reconstruction. Osteotomy complications are rare and often self-limiting, but include increased risk of transient nerve and muscle palsies (typically resolve within 12 weeks of surgery), delayed ileal union, and superficial pin-site infection or inflammation.59 In the experience at the authors’ institution, even though pelvic osteotomy with postoperative immobilization may extend hospital stay, the drastic reduction in failure rate is considered worthwhile by patients and care providers.
Emerging evidence suggests the timing of the initial closure impacts success rates. In 2014, Shah and colleagues reported outcomes in 60 patients with CE and found that a 1 cm increase in pre-closure diastasis resulted in a 2.64 increase in the odds of failed initial closure. Subsequently performing osteotomy and delaying closure while allowing the external fixator to decrease the diastasis increased closure success. Interestingly, 77% of patients with a failed closure were closed within the first week of life compared to 26% of patients who underwent staged closure. Additionally, only 31% of patients in the failed closure group underwent osteotomy during initial closure group compared to 82% of the staged closure patients. Other protective strategies against failure include delaying initial bladder closure until after omphalocele closure.60 Finally, 92% of patients with a successful re-closure underwent osteotomy.47 These results demonstrating increased success in delayed primary repair were supported in another series that demonstrated a higher percentage of failures in primary bladder closure in patients closed within the first 30 days of life compared to those performed after 30 days of age (47.7% vs 19.3%).55
To achieve continence after a failed primary closure, a secondary bladder closure must be performed. Similar to findings reported by Shah et al., Davis and colleagues found that the age at which the secondary bladder closure is performed is predictive of overall success. They reported patients with a successful secondary bladder closure were more than a year older than patients who failed a secondary bladder closure (median age 104 weeks vs 38 weeks).56 Successful secondary closure had a median delay of 154 weeks compared to a median delay of 30 weeks in failed secondary closures. Failed urinary reconstruction in children with exstrophy is significant as it may lead to blunted bladder growth and loss of the bladder template, which may make it increasingly difficult, if not impossible, to achieve continence.
Achieving Continence
Due to the frequent conglomeration of possible anomalies—small bladder templates, poorly compliant bladders, widely patent bladder neck (if at all present), and spinal anomalies that contribute to neurogenic bowel and bladder—voided continence is unlikely. However, continence remains the ultimate goal in exstrophy reconstruction to enable these children to live more typical lives. To this end, additional procedures are often required, as confirmed by Maruf et al in a large series that reported that patients require a median of 2 (range 1–4) urinary continence procedures to achieve a dry interval greater than 3 hours.61
In a large series from the authors’ institution assessing outcomes in 80 patients with OEIS complex, data on urinary reconstruction was available for 73 patients. Of the 40 dry patients, 38 retained their native bladder while 2 had small bowel neobladders (Koch pouch). Of the 38 patients with a native bladder, 30 (79%) received a continent catheterizable channel (continent urinary diversion, CUD) with either appendix (n =3) or bowel segment (n = 27), and 32 (84%) also had augmentation cystoplasty (AC) prior to achieving dryness. Only 9 patients (24%) maintained a patent and continent urethra.46 In an aggregate of studies assessing urinary continence in CE, AC was required in up to 50% of patients and an overall dry rate of 72%.62
These additional adjuncts for continence, AC and CUD, present unique complications including mucus overproduction, bladder calculi, chronic bacterial colonization, and epithelial polyps. Other long-term issues arise from AC including chronic metabolic acidosis and, less likely, carcinoma.63,64 With time, the stoma created for CUD may stenose, prolapse, necrose, or leak, requiring revision.64
In genetic females, successful continence may be achieved after Young-Dees-Leadbetter BNR, though most patients require clean intermittent catheterization. Techniques utilized in both males and females to enhance bladder capacity were previously described but include incorporating the hindgut segment (when available), the small and large bowels, and, less commonly, the stomach.38 Achieving continence is much more challenging in the reassigned male-to-female patient and a continent stoma may be the most advantageous strategy in the long-term.17 Regardless of the techniques employed, in our institution’s experience, children are continent of urine at a median age of 11 years.61 In one large series evaluating continence in cloacal exstrophy patients 50% (35 of 61) of children achieved continence, with 30 of 35 intermittently catheterizing via a continent stoma and the remainder voiding or catheterizing per urethra.65
Suggested Follow-Up
Patients with exstrophy, their families, and the treating medical and surgical teams develop a special bond that is unique in medicine. Following urinary tract and abdominal wall reconstruction and possible subsequent epispadias repair, the focus in management shifts to protecting the upper urinary tracts. To this end, at the author’s institution, we monitor for any evidence of reflux with yearly cystograms and cystoscopy while tracking bladder growth. In school-aged children (at least 5-7 years old) we assess bladder capacity, urodynamics, and the child’s ability to participate in their own care, through the evaluation of dexterity and maturity, prior to bladder neck reconstruction.31
Conclusions
CE is a devastating multisystem birth defect that once predisposed children to significant morbidity and even mortality. Great strides, however, in the surgical and medical management of these children have enabled them to live near normal lives into adulthood. Success in contemporary management stems from the multidisciplinary approach to overcoming the challenges these patients face. As these patients continue to live healthy lives well into adulthood, we will continue to learn more with regards to quality of life and ways in which overall patient function may be optimized.
Key Points
- Accurate and thorough perinatal diagnosis is crucial in organizing appropriate multidisciplinary care—fUS and fMRI are useful modalities in this endeavor.
- Initial postnatal management should ensure the patient is medically stable. Once stable, a thorough physical exam and proper imaging should be utilized to ensure complete recognition of all associated anomalies.
- Abdominal and urinary tract reconstruction can occur only after neurosurgical evaluation for spine or CNS involvement and clearance.
- Any hindgut remnant should be preserved at all costs in these children as it may be used in gastrointestinal or genitourinary reconstruction.
- Separation of the bladder from the gastrointestinal tract with concurrent fecal diversion is key prior to future reconstruction. End-colostomy has emerged as the least morbid form of fecal diversion.
- Close follow-up after reconstruction in these patients is essential to ensure preservation of upper tract function.
Future Directions
Current research seeks to improve counseling and decrease morbidity associated with surgery by developing better predictive tools for patients and families. Better counseling may be achieved through tools, such as ones that predict bladder capacity and growth (Sholklapper et al 2022, submitted), which may have implications on future surgery and ability to achieve continence.66 Additionally, basic science research evaluating ultrastructure differences between exstrophy bladders and normal age-matched controls may provide a clearer picture of differential bladder growth curves for these patients. The use of imaging to assess pelvic floor anatomy as a tool for surgical planning provides additional information for intraoperative decision making and the use of this imaging is expected to expand. Long-term, addressing the growing field of translational urology and who cares for these children as they enter adulthood will help optimize care for these patients. Finally, the expansion of telemedicine for consultation among medical centers may improve communication regarding this rare condition and improve patient and family wellness through reduced burden related to accessibility, travel, and cost expenditures.
Suggested Readings
- Gearhart JP, Carlo HN. Exstrophy-epispadias complex. 12th ed., Philadelphia, PA: Elsevier; 2020, DOI: 10.1201/9780429194993-25.
- Grady RW, Mitchell ME. Complete Primary Repair Of Exstrophy. J Urol 1999; 65 (1): 1415–1420. DOI: 10.1097/00005392-199910000-00071.
- Davis R, Stewart D, Maruf M, Lau H, Gearhart JP. Complex abdominal wall reconstruction combined with bladder closure in OEIS complex. J Pediatr Surg 2019; 54 (11): 2408–2412. DOI: 10.1016/j.jpedsurg.2019.03.022.
- Tourchi A, Inouye BM, Di Carlo HN, Young E, Ko J, Gearhart JP. New advances in the pathophysiologic and radiologic basis of the exstrophy spectrum. Journal of Pediatric Urology 2014; 10 (2): 212–218.
- Kasprenski M, Michaud J, Yang Z, Maruf M, Benz K, Jayman J, et al.. Urothelial differences in the exstrophy-epispadias complex: potential implications for management. The Journal of Urology 2021; 205 (5): 1460–1465.
- Di Carlo HN, Maruf M, Massanyi EZ, Shah B, Tekes A, Gearhart JP. 3-dimensional magnetic resonance imaging guided pelvic floor dissection for bladder exstrophy: a single arm trial. The Journal of Urology 2019; 202 (2): 406–412.
References
- Muecke EC. The Role of the Cloacae Membrane in Exstrophy: The First Successful Experimental Study. J Urol 1069; 92 (6): 659–668. DOI: 10.1016/s0022-5347(17)64028-x.
- Woo LL, Thomas JC, Brock JW. Cloacal exstrophy: A comprehensive review of an uncommon problem. J Pediatr Urol 2010; 6 (2): 102–111. DOI: 10.1016/j.jpurol.2009.09.011.
- Moore KL. Organogenetic Period: The Fourth to Eighth Weeks. In: Moore KL, Persaud TVN, editors. The Developing Human: Clinically Oriented Embryology. 7th ed. Philadelphia, PA: Saunders; 2003.
- Howell C, Caldamone A, Snyder H, Ziegler M, Duckett J. Optimal management of cloacal exstrophy. J Pediatr Surg 1983; 18 (4): 365–369. DOI: 10.1016/s0022-3468(83)80182-1.
- Cervellione RM, Mantovani A, Gearhart J, Bogaert G, Gobet R, Caione P, et al.. Prospective study on the incidence of bladder/cloacal exstrophy and epispadias in Europe. J Pediatr Urol 2015; 11 (6): 337.e1–337.e6. DOI: 10.1016/j.jpurol.2015.03.023.
- Shapiro E, Lepor H, Jeffs RD. The Inheritance of the Exstrophy-Epispadias Complex. J Urol 1984; 132 (2): 308–310. DOI: 10.1016/s0022-5347(17)49605-4.
- Ives E, Coffey R, Carter CO. A family study of bladder exstrophy. J Med Genet 1980; 17 (2): 139–141. DOI: 10.1136/jmg.17.2.139.
- Birth Defects Monitoring Systems IC for. Epidemiology of bladder exstrophy and epispadias: A communication from the international clearinghouse for birth defects monitoring systems. Teratology 1987; 36 (2): 221–227. DOI: 10.1002/tera.1420360210.
- Botto LD, Feldkamp ML, Amar E, Carey JC, Castilla EE, Clementi M, et al.. Acardia: Epidemiologic findings and literature review from the International Clearinghouse for Birth Defects Surveillance and Research. Am J Med Genet C Semin Med Genet 2011; 157 (4): 262–273. DOI: 10.1002/ajmg.c.30318.
- Boyadjiev SA, Dodson JL, Radford CL, Ashrafi GH, Beaty TH, Mathews RI, et al.. Clinical and molecular characterization of the bladder exstrophy-epispadias complex: analysis of 232 families. BJU Int 2004; 94 (9): 1337–1343. DOI: 10.1111/j.1464-410x.2004.05170.x.
- WOOD HADLEYM, TROCK BRUCEJ, GEARHART JOHNP. In Vitro Fertilization and the Cloacal-Bladder Exstrophy-Epispadias Complex: Is there an Association? J Urol 2003; 169 (4): 1512–1515. DOI: 10.1097/01.ju.0000054984.76384.66.
- Reutter H, Boyadjiev SA, Gambhir L, Ebert A-K, Rösch WH, Stein R, et al.. Phenotype Severity in the Bladder Exstrophy-Epispadias Complex: Analysis of Genetic and Nongenetic Contributing Factors in 441 Families from North America and Europe. J Pediatr 2011; 159 (5): 825–831.e1. DOI: 10.1016/j.jpeds.2011.04.042.
- Sponseller PD, Bisson LJ, Gearhart JP, Jeffs RD, Magid D, Fishman E. The anatomy of the pelvis in the exstrophy complex. J Bone Joint Surg Am 1995; 77 (2): 177–189. DOI: 10.2106/00004623-199502000-00003.
- Stec AA, Pannu HK, Tadros YE, Sponseller PD, Fishman EK, Gearhart JP. Pelvic Floor Anatomy In Classic Bladder Exstrophy Using 3-dimensional Computerized Tomography: J Urol 2001; 166 (4): 1444–1449. DOI: 10.1097/00005392-200110000-00066.
- Greene WB, Dias LS, Lindseth RE, Torch MA. Musculoskeletal problems in association with cloacal exstrophy. J Bone Joint Surg Am 1991; 73 (4): 551–560. DOI: 10.2106/00004623-199173040-00012.
- Davidoff AM, Hebra A, Balmer D, Templeton JM, Schnaufer L. Management of the gastrointestinal tract and nutrition in patients with cloacal exstrophy. J Pediatr Surg 1996; 31 (6): 771–773. DOI: 10.1016/s0022-3468(96)90129-3.
- Mathews R, Jeffs RD, Reiner WG, Docimo SG, Gearhart JP. Cloacal Exstrophy-improving The Quality Of Life. J Urol 1998; 6 (2): 2452–2456. DOI: 10.1097/00005392-199812020-00017.
- McHoney M, Ransley PG, Duffy P, Wilcox DT, Spitz L. Cloacal exstrophy: Morbidity associated with abnormalities of the gastrointestinal tract and spine. J Pediatr Surg 2004; 39 (8): 1209–1213. DOI: 10.1016/j.jpedsurg.2004.04.019.
- Sugar EC, Firlit CF. Management of cloacal exstrophy. Urology 2004; 32 (4): 320–322. DOI: 10.1016/0090-4295(88)90234-8.
- Sawaya D, Goldstein S, Seetharamaiah R, Suson K, Nabaweesi R, Colombani P, et al.. Gastrointestinal ramifications of the cloacal exstrophy complex: a 44-year experience. J Pediatr Surg 2010; 45 (1): 171–176. DOI: 10.1016/j.jpedsurg.2009.10.030.
- Hurwitz RS, Manzoni GAM, Ransley PG, Stephens FD. Cloacal Exstrophy: A Report of 34 Cases. J Urol 1987; 138 (4 Part 2): 1060–1064. DOI: 10.1016/s0022-5347(17)43502-6.
- Phillips TM, Salmasi AH, Stec A, Novak T, Gearhart JP, Mathew J. Re: Urological Outcomes in the Omphalocele Exstrophy Imperforate Anus Spinal Defects (OEIS) Complex: Experience with 80 Patients. J Urol 2013; 191 (4): 1118–1119. DOI: 10.1016/j.juro.2014.01.044.
- Schlegel PN, Gearhart JP. Neuroanatomy of the Pelvis in an Infant with Cloacal Exstrophy: A Detailed Microdissection with Histology. J Urol 1989; 141 (3 Part 1): 583–585. DOI: 10.1016/s0022-5347(17)40901-3.
- Connolly JA, Peppas DS, Jeffs RD, Gearhart JP. Prevalence and Repair of Inguinal Hernias in Children with Bladder Exstrophy. J Urol 1995; 154 (5): 1900–1901. DOI: 10.1097/00005392-199511000-00093.
- Sadula SR, Kanhere SV, Phadke VD. Exstrophy of Cloaca Sequence (Oeis Complex) With Multiple Cardiac Malformations. Indian Journal of Case Reports 2019; 05 (04): 317–319. DOI: 10.32677/ijcr.2019.v05.i04.006.
- Meizner I, Bar-Ziv J. In utero prenatal ultrasonic diagnosis of a rare case of cloacal exstrophy. J Clin Ultrasound 1985; 13 (7): 500–502. DOI: 10.1002/jcu.1870130714.
- Austin PF, Homsy YL, Gearhart JP, Porter K, Guidi C, Madsen K, et al.. The Prenatal Diagnosis Of Cloacal Exstrophy. J Urol 1998; 160 (3.2): 1179–1181. DOI: 10.1097/00005392-199809020-00061.
- Calvo-Garcia MA, Kline-Fath BM, Rubio EI, Merrow AC, Guimaraes CV, Lim F-Y. Fetal MRI of cloacal exstrophy. Pediatr Radiol 2013; 43 (5): 593–604. DOI: 10.1007/s00247-012-2571-3.
- Goto S, Suzumori N, Obayashi S, Mizutani E, Hayashi Y, Sugiura-Ogasawara M. Prenatal findings of omphalocele-exstrophy of the bladder-imperforate anus-spinal defects (OEIS) complex. Congenit Anom (Kyoto) 2012; 52 (3): 179–181. DOI: 10.1111/j.1741-4520.2011.00342.x.
- Weiss DA, Oliver ER, Borer JG, Kryger JV, Roth EB, Groth TW, et al.. Key anatomic findings on fetal ultrasound and MRI in the prenatal diagnosis of bladder and cloacal exstrophy. J Pediatr Urol 2020; 16 (5): 665–671. DOI: 10.1016/j.jpurol.2020.07.024.
- Lowentritt BH, Van Zijl PS, Frimberger D, Baird A, Lakshmanan Y, Gearhart JP. Variants Of The Exstrophy Complex: A Single Institution Experience. J Urol 2005; 173 (5): 1732–1737. DOI: 10.1097/01.ju.0000154353.03056.5c.
- Gearhart JP, Jeffs RD. The bladder exstrophy-epispadias complex. 3rd ed., Philadelphia, PA: Saunders; 1998, DOI: 10.1007/978-3-319-44182-5_13.
- Tank ES, Lindenauer SM. Principles of management of exstrophy of the cloaca. Am J Surg 1970; 119 (1): 95–98. DOI: 10.1016/0002-9610(70)90018-8.
- Reiner WG, Gearhart JP. Discordant Sexual Identity in Some Genetic Males with Cloacal Exstrophy Assigned to Female Sex at Birth. N Engl J Med 2004; 350 (4): 333–341. DOI: 10.1056/nejmoa022236.
- Baker Towell DM, Towell AD. A Preliminary Investigation Into Quality of Life, Psychological Distress and Social Competence in Children With Cloacal Exstrophy. J Urol 2003; 169 (5): 1850–1853. DOI: 10.1097/01.ju.0000062480.01456.34.
- Mirheydar H, Evason K, Coakley F, Baskin LS, DiSandro M. 46, XY female with cloacal exstrophy and masculinization at puberty. J Pediatr Urol 2009; 5 (5): 408–411. DOI: 10.1016/j.jpurol.2009.03.013.
- Diamond DA, Burns JP, Mitchell C, Lamb K, Kartashov AI, Retik AB. Sex assignment for newborns with ambiguous genitalia and exposure to fetal testosterone: Attitudes and practices of pediatric urologists. J Pediatr 2006; 148 (4): 445–449. DOI: 10.1016/j.jpeds.2005.10.043.
- Fullerton BS, Sparks EA, Hall AM, Chan Y-M, Duggan C, Lund DP, et al.. Growth morbidity in patients with cloacal exstrophy: a 42-year experience. J Pediatr Surg 2016; 51 (6): 1017–1021. DOI: 10.1016/j.jpedsurg.2016.02.075.
- Gearhart JP, Jeffs RD. Techniques to Create Urinary Continence in the Cloacal Exstrophy Patient. J Urol 1991; 146 (2 Part 2): 616–618. DOI: 10.1016/s0022-5347(17)37871-0.
- Davis R, Stewart D, Maruf M, Lau H, Gearhart JP. Complex abdominal wall reconstruction combined with bladder closure in OEIS complex. J Pediatr Surg 2019; 54 (11): 2408–2412. DOI: 10.1016/j.jpedsurg.2019.03.022.
- Henderson CG, North AC, Gearhart JP. The use of alloderm as an adjunct in the closure of the bladder – Cloacal exstrophy complex. J Pediatr Urol 2011; 7 (1): 44–47. DOI: 10.1016/j.jpurol.2010.02.209.
- Mathews, Perlman, Marsh, Gearhart. Gonadal morphology in cloacal exstrophy: implications in gender assignment. BJU Int 1999; 84 (1): 99–100. DOI: 10.1046/j.1464-410x.1999.00148.x.
- Lumen N, Monstrey S, Selvaggi G, Ceulemans P, De Cuypere G, Van Laecke E, et al.. Phalloplasty: A Valuable Treatment for Males with Penile Insufficiency. Urology 2008; 71 (2): 272–276. DOI: 10.1016/j.urology.2007.08.066.
- Gearhart JP, Leonard MP, Burgers JK, Jeffs RD. The Cantwell-Ransley Technique for Repair of Epispadias. J Urol 1992; 148 (3 Part 1): 851–854. DOI: 10.1016/s0022-5347(17)36742-3.
- Gearhart JP, Carlo HN. Exstrophy-epispadias complex. 12th ed., Philadelphia, PA: Elsevier; 2020, DOI: 10.1201/9780429194993-25.
- Ben-Chaim J, Peppas DS, Sponseller PD, Jeffs RD, Gearhart JP. Applications of Osteotomy in the Cloacal Exstrophy Patient. J Urol 1995; 154 (2.2): 865–867. DOI: 10.1097/00005392-199508000-00146.
- Mathews R, Gearhart JP, Bhatnagar R, Sponseller P. Staged Pelvic Closure of Extreme Pubic Diastasis in the Exstrophy-Epispadias Complex. J Urol 2006; 176 (5): 2196–2198. DOI: 10.1016/j.juro.2006.07.058.
- Benz KS, Jayman J, Maruf M, Baumgartner T, Kasprenski MC, Friedlander DA, et al.. Pelvic and lower extremity immobilization for cloacal exstrophy bladder and abdominal closure in neonates and older children. J Pediatr Surg 2018; 53 (11): 2160–2163. DOI: 10.1016/j.jpedsurg.2017.11.066.
- Grady RW, Mitchell ME. Complete Primary Repair Of Exstrophy. J Urol 1999; 65 (1): 1415–1420. DOI: 10.1097/00005392-199910000-00071.
- Thomas JC, DeMarco RT, Pope JC, Adams MC, Brock JW. First Stage Approximation of the Exstrophic Bladder in Patients With Cloacal Exstrophy–Should This be the Initial Surgical Approach in all Patients? J Urol 2007; 178 (4s): 1632–1636. DOI: 10.1016/j.juro.2007.03.164.
- Lee RS, Grady R, Joyner B, Casale P, Mitchell M. Can a Complete Primary Repair Approach be Applied to Cloacal Exstrophy? J Urol 2006; 176 (6): 2643–2648. DOI: 10.1016/j.juro.2006.08.052.
- Mitchell ME, Bagli DJ. Complete Penile Disassembly for Epispadias Repair. J Urol 1996; 155 (1): 300–304. DOI: 10.1097/00005392-199601000-00128.
- Gargollo P, Hendren WH, Diamond DA, Pennison M, Grant R, Rosoklija I, et al.. Bladder Neck Reconstruction is Often Necessary After Complete Primary Repair of Exstrophy. J Urol 2011; 185 (6s): 2563–2571. DOI: 10.1016/j.juro.2011.01.024.
- Schaeffer AJ, Stec AA, Purves JT, Cervellione RM, Nelson CP, Gearhart JP. Complete Primary Repair of Bladder Exstrophy: A Single Institution Referral Experience. J Urol 2011; 186 (3): 1041–1047. DOI: 10.1016/j.juro.2011.04.099.
- Friedlander DA, Di Carlo HN, Sponseller PD, Gearhart JP. Complications of bladder closure in cloacal exstrophy: Do osteotomy and reoperative closure factor in? J Pediatr Surg 2017; 52 (11): 1836–1841. DOI: 10.1016/j.jpedsurg.2016.12.002.
- Davis R, Sood A, Maruf M, Singh P, Kasprenski MC, DiCarlo HN, et al.. The failed bladder closure in cloacal exstrophy: Management and outcomes. J Pediatr Surg 2019; 54 (11): 2416–2420. DOI: 10.1016/j.jpedsurg.2019.02.012.
- Goldstein SD, Inouye BM, Reddy S, Lue K, Young EE, Abdelwahab M, et al.. Continence in the cloacal exstrophy patient: What does it cost? J Pediatr Surg 2016; 51 (4): 622–625. DOI: 10.1016/j.jpedsurg.2015.12.003.
- Jayman J, Tourchi A, Feng Z, Trock BJ, Maruf M, Benz K, et al.. Predictors of a successful primary bladder closure in cloacal exstrophy: A multivariable analysis. J Pediatr Surg 2019; 54 (3): 491–494. DOI: 10.1016/j.jpedsurg.2018.06.030.
- Inouye BM, Tourchi A, Di Carlo HN, Young EE, Gearhart JP. Modern Management of the Exstrophy-Epispadias Complex. Surg Res Pract 2014; 2014 (587064): 1–9. DOI: 10.1155/2014/587064.
- Shah BB, Di Carlo H, Goldstein SD, Pierorazio PM, Inouye BM, Massanyi EZ, et al.. Initial bladder closure of the cloacal exstrophy complex: Outcome related risk factors and keys to success. J Pediatr Surg 2014; 49 (6): 1036–1040. DOI: 10.1016/j.jpedsurg.2014.01.047.
- Maruf M, Kasprenski M, Jayman J, Goldstein SD, Benz K, Baumgartner T, et al.. Achieving urinary continence in cloacal exstrophy: The surgical cost. J Pediatr Surg 2018; 53 (10): 1937–1941. DOI: 10.1016/j.jpedsurg.2018.02.055.
- Mathews R. Achieving urinary continence in cloacal exstrophy. Semin Pediatr Surg 2011; 20 (2): 126–129. DOI: 10.1053/j.sempedsurg.2010.12.009.
- Surer I, Ferrer FA, Baker LA, Gearhart JP. Continent Urinary Diversion and the Exstrophy-Epispadias Complex. J Urol 2003; 169 (3): 1102–1105. DOI: 10.1097/01.ju.0000044921.19074.d0.
- Woodhouse CRJ, North AC, Gearhart JP. Standing the test of time: long-term outcome of reconstruction of the exstrophy bladder. World J Urol 2006; 24 (3): 244–249. DOI: 10.1007/s00345-006-0053-7.
- Suson K, Novak T, Gupta A, Benson J, Sponseller PD, Gearhart JP. The Neuro-Orthopedic Manifestations of the Omphalocele Exstrophy Imperforate Anus Spinal Defects (OEIS) Complex. J Pediatr Urol 2010; 5 (4): S54. DOI: 10.1016/j.jpurol.2009.02.083.
- Di Carlo HN, Maruf M, Massanyi EZ, Shah B, Tekes A, Gearhart JP. 3-dimensional magnetic resonance imaging guided pelvic floor dissection for bladder exstrophy: a single arm trial. The Journal of Urology 2019; 202 (2): 406–412.
- Tourchi A, Inouye BM, Di Carlo HN, Young E, Ko J, Gearhart JP. New advances in the pathophysiologic and radiologic basis of the exstrophy spectrum. Journal of Pediatric Urology 2014; 10 (2): 212–218.
- Kasprenski M, Michaud J, Yang Z, Maruf M, Benz K, Jayman J, et al.. Urothelial differences in the exstrophy-epispadias complex: potential implications for management. The Journal of Urology 2021; 205 (5): 1460–1465.
Last updated: 2025-09-25 12:10