
22: Extrofia cloacal
Este capítulo durará aproximadamente 35 minutos para leer.
Introducción
La extrofia cloacal (CE), comúnmente denominada complejo de onfalocele, extrofia vesical, ano imperforado y anomalías espinales (OEIS), es la manifestación menos frecuente del complejo extrofia-epispadias (EEC); sin embargo, representa la mayor desviación respecto de la anatomía y el desarrollo típicos. Como defecto multisistémico, requiere un enfoque multidisciplinario dedicado para lograr resultados óptimos para los pacientes y sus familias. Este capítulo ofrece una revisión de la extrofia cloacal desde una perspectiva moderna, junto con direcciones futuras para mejorar la comprensión de esta afección que supone una gran carga.
Embriología
La teoría predominante detrás de la EEC es el fracaso de la interposición del mesodermo para reforzar la membrana cloacal.1 La membrana cloacal es una capa bilaminar de tejido ectodérmico y endodérmico situada en el aspecto caudal del disco germinal de la pared abdominal infraumbilical en desarrollo. En el desarrollo normal, durante la 4.a y 5.a semanas de gestación, el crecimiento del mesénquima entre estas capas forma los músculos abdominales inferiores y la pelvis ósea. El descenso caudal continuado de este tabique urorectal da lugar a la fusión con la membrana cloacal y, en última instancia, a la separación de la cloaca en vejiga anteriormente y recto posteriormente (Figura 1).2 La perforación de la membrana cloacal suele ocurrir tras la fusión con el tabique urorectal, aproximadamente en la 6.a semana del desarrollo, formando orificios urogenital y anal separados. Los tubérculos genitales pares que dan origen al falo, luego migran medialmente y se fusionan en la línea media.3
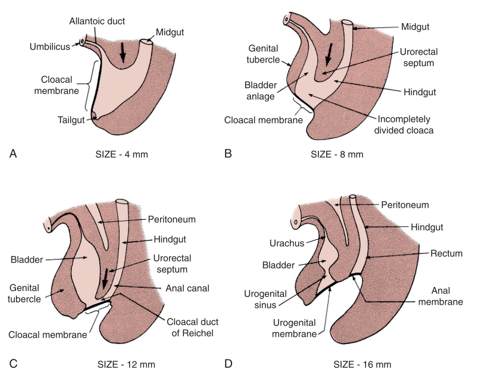
Figura 1 Desarrollo de la membrana cloacal, el seno urogenital y las aberturas anales. Crecimiento del mesénquima con descenso caudal del tabique urorrectal y perforación de la membrana cloacal, lo que da lugar a las cavidades urogenital y anal.
La falta de refuerzo mesenquimático y el desarrollo excesivo resultante de la membrana cloacal conllevan riesgo de rotura prematura de la membrana cloacal. El momento y el grado en que se produce la rotura prematura de esta membrana dan lugar al espectro de anomalías observado en la EEC; la CE es el defecto de formación más precoz.
Epidemiología
La incidencia de la CE es de aproximadamente 1 de cada 200,000-400,000 nacidos vivos4,5 Históricamente, en la CE existe un predominio masculino:femenino de 2:1, aunque algunas series sitúan esta relación más cerca de 1:1.6,7,8 Sin embargo, con la mejora de las modalidades de imagen y el diagnóstico prenatal, el número de nacidos vivos con CE ha disminuido de manera marcada, ya que los embarazos se interrumpen de manera electiva en el 23% de los casos y hasta el 50% de los embarazos terminan en aborto espontáneo.9
La mayoría de los casos parecen esporádicos; sin embargo, los análisis genéticos han identificado posibles factores causales. Thauvin-Robinet et al describieron una translocación desequilibrada entre el brazo largo del cromosoma 9 y el cromosoma Y que produce una deleción 9q34.1-qter como posible causa, mientras que otros implicaron mutaciones en un grupo de genes homeobox, incluidos HLXB9 y la familia HOX, que afectan el desarrollo mesodérmico.10
Además de los antecedentes familiares, se han implicado otros factores de riesgo específicos de la CE. En un análisis clínico y molecular de 232 familias, la edad parental avanzada emergió como un posible factor de riesgo, con una edad materna promedio de 34 años y una edad paterna promedio de 32 años (en comparación con 26 y 27 años según los datos del censo de 2010, respectivamente). Curiosamente, el 49 % de los casos de exstrofia se produjeron en primeros embarazos, lo que también puede reforzar la asociación con la mayor edad parental.10 Las influencias hormonales también pueden desempeñar un papel, ya que Wood y colegas identificaron un aumento de 10 veces en los nacimientos de niños con exstrofia entre madres que recibieron altas dosis de progesterona al inicio del primer trimestre.11 En este mismo estudio, también se informaron asociaciones con las técnicas de reproducción asistida, ya que los niños concebidos mediante fertilización in vitro (FIV) presentaron una incidencia de exstrofia 7,5 veces mayor.
Actualmente, el factor ambiental más estrechamente asociado con la extrofia cloacal es la exposición materna al tabaco, específicamente la exposición materna al tabaquismo en el periodo periconcepcional y el tabaquismo materno durante el primer trimestre.12 En conjunto, surgen dos puntos críticos en el asesoramiento: 1) el asesoramiento adecuado sobre la optimización de los periodos periconcepcional y del primer trimestre – durante el tiempo de la organogénesis y 2) la comprensión de los cambios en las perspectivas sociales hacia la planificación familiar (con padres primerizos de mayor edad) y la posible necesidad de tecnologías de reproducción asistida.
Patogénesis y anomalías asociadas
La complejidad para determinar las causas multifactoriales subyacentes a la CE se demuestra en la variedad de defectos observados en este fenotipo (Tabla 1). Dado que la CE es el resultado de eventos del desarrollo temprano, se presenta con más anomalías y en el grado más severo, y representa el extremo más complejo del espectro de extrofia-epispadias. La expresión fenotípica clásica de la CE involucra una franja de ciego extrofiado flanqueada por dos mitades vesicales extrofiadas, que a menudo se acompañan de un segmento ileal prolapsado que confiere la deformidad de “trompa de elefante” (Figura 2). Además, el intestino posterior está acortado y a menudo termina en fondo de saco, lo que da lugar a un ano imperforado. Como los tubérculos genitales no logran fusionarse en la línea media, se observan dos mitades fálicas a cada lado de una diástasis púbica ensanchada (Figura 3). El defecto de la pared abdominal generalmente se acompaña de un onfalocele de tamaño variable.2 Debido a la naturaleza multisistémica de este defecto, cada anomalía se describe con más detalle por separado.
Tabla 1 Extrofia cloacal y sus anomalías asociadas.
| Gastrointestinal | Genitourinario | Sistema nervioso central | Musculoesquelético |
|---|---|---|---|
| Onfalocele | Agenesia renal unilateral | Médula anclada | Vertebrales (ausencia o hemivértebras) |
| Ano imperforado, atresia o estenosis anal | Riñón pélvico | Mielomeningocele | Pie equinovaro |
| Síndrome de intestino corto | Duplicación ureteral | Subluxación de cadera | |
| Malrotación intestinal | Hidronefrosis | Escasez de musculatura del suelo pélvico anterior | |
| Duplicación intestinal | Criptorquidia bilateral | ||
| Hernias inguinales | |||
| Duplicación uterina | |||
| Duplicación vaginal |
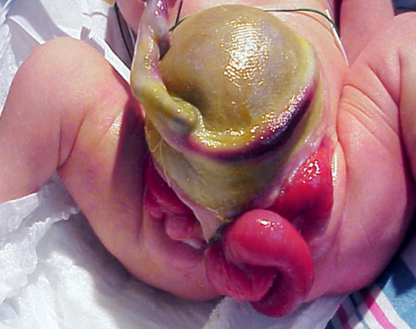
Figura 2 Varón con presentación clásica de extrofia cloacal con segmento ileal prolapsado que crea la deformidad de “trompa de elefante”.
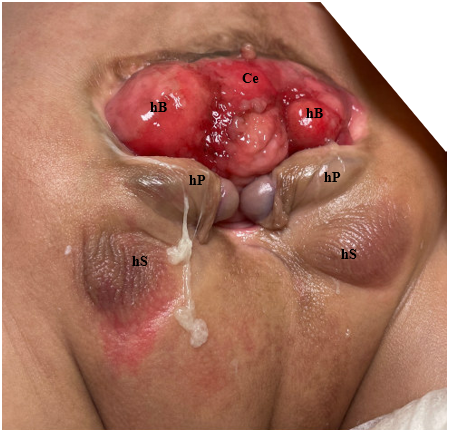
Figura 3 Fotografía de un varón con extrofia cloacal. Obsérvese que ya se ha realizado una colostomía. Ce = ciego; hB = hemivejiga; hP = hemifalo; hS = hemiescroto.
Musculoesquelético
Pelvis ósea
Las anomalías de la pelvis ósea y la diástasis púbica asociada son el sello distintivo de la extrofia. En 1995, Sponseller et al utilizaron tomografía computarizada (TC) tridimensional (3D) de la pelvis ósea en pacientes con extrofia y elucidaron anomalías que modernizaron nuestra comprensión del ensanchamiento característico de la sínfisis púbica. Lo más notable es que identificaron dos categorías amplias de anomalías, consideradas anomalías rotacionales y dimensionales (Tabla 2).
Tabla 2 Anomalías de la pelvis ósea.
| Rotacional | Dimensional |
|---|---|
| Rotación externa de la pelvis posterior/alas ilíacas | Aumento de la diástasis púbica |
| Rotación externa del segmento pélvico anterior | Acortamiento del segmento púbico anterior (30%) |
| Rotación coronal de la articulación sacroilíaca | Aumento de la distancia entre los cartílagos trirradiados |
| Retroversión acetabular | |
| Convergencia de las alas ilíacas | |
| Retroversión femoral |
En comparación con controles emparejados por edad, Sponseller et al encontraron que, en la extrofia vesical clásica (CBE), la pelvis posterior está rotada externamente un promedio de 12 grados en cada lado, con la pelvis anterior rotada externamente un promedio de 18 grados. Además, las ramas púbicas son un 30% más cortas, lo cual, combinado con un acetábulo retrovertido, conduce a una diástasis promedio de 4.8 cm en la CBE (Figura 4).13 En los pacientes con CE, estos defectos son aún más acentuados, con una disminución global del 43% en la longitud ósea de toda la pelvis, con una diástasis púbica media mayor de 6 cm y una mayor probabilidad de asimetría entre los lados derecho e izquierdo de la pelvis. Para mayor claridad, una diástasis leve es inferior a 4 cm, una diástasis moderada está entre 4 y 6 cm y una diástasis extrema es cualquier valor > 6 cm.
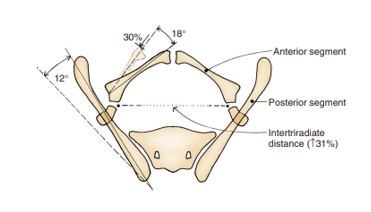
Figura 4 Anomalías óseas pélvicas observadas en la extrofia vesical clásica. El segmento óseo posterior está rotado externamente 12˚ a cada lado, pero la longitud no se modifica. El segmento anterior está rotado externamente 18˚ a cada lado y acortado en un 30%. La distancia entre los cartílagos trirradiados se incrementa en un 31%. Obsérvese que las anomalías son más graves en la extrofia cloacal y la asimetría es más frecuente. Utilizado con permiso del Brady Urological Institute.
Defectos del suelo pélvico
Las anomalías rotacionales y dimensionales de la pelvis ósea impactan directamente en el maldesarrollo del suelo pélvico. Las imágenes de TC en 3D de niños con extrofia revelaron que los músculos elevadores del ano están posicionados más posteriormente (68% posteriormente/32% anteriormente) en comparación con controles pareados por edad (52% posteriormente/48% anteriormente) (Figura 5).14 Además de estar orientado más posteriormente, el elevador del ano también es menos cóncavo. Otra consecuencia de las deformidades de la pelvis ósea manifestadas en el suelo pélvico es un músculo puborrectal más aplanado en comparación con su forma cónica habitual. La escasez de musculatura anterior del suelo pélvico y la falta de una forma cónica orientan la técnica quirúrgica en la reconstrucción de la pelvis ósea y el suelo pélvico, lo cual se analiza por separado.
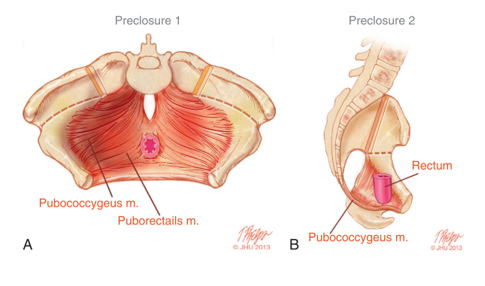
Figura 5 A) Musculatura del suelo pélvico previa al cierre de la extrofia con las incisiones de osteotomía marcadas. B) Vista lateral de la anatomía del suelo pélvico que muestra el desplazamiento posterior de los músculos del suelo pélvico por detrás del recto.
Alteraciones de las extremidades inferiores y de la marcha
Las anomalías de las extremidades inferiores se observan de forma constante en la CE. Como resultado del aumento de la diástasis púbica, la distancia entre las caderas y la rotación externa de los acetábulos, los niños pueden compensar con rotación externa de los miembros inferiores y una marcha bamboleante. Las deformidades adicionales incluyen torsión tibial, deformidades equinovaras y calcáneas.15 A pesar de estas deformidades de las extremidades, el rango de movimiento en estos pacientes por lo general no se ve afectado y las órtesis correctoras según sea necesario ayudan a corregir la marcha con el tiempo. Para los 2 años de edad, la mayoría de los niños deambulan con un uso mínimo de dispositivos de asistencia y pocos requieren el uso de silla de ruedas durante la infancia.
Anomalías gastrointestinales
Las anomalías gastrointestinales se presentan en cierta medida en casi todos los pacientes y contribuyen de manera significativa a la morbilidad y al grado de dificultad en el manejo de estos pacientes, tanto en el ámbito perioperatorio en lo referente a la nutrición como a largo plazo. El onfalocele es característico de esta afección y se encuentra en el 88–100% de los casos.16,17,18,19 Los onfaloceles pueden variar ampliamente en tamaño y en su contenido, conteniendo intestino delgado, hígado o ambos. El cierre inmediato del defecto del onfalocele o su colocación en un silo protector durante el periodo neonatal suele realizarse para prevenir una rotura posterior y proporcionar un entorno propicio. En una serie de Davidoff et al que incluyó a 26 pacientes con CE, todos presentaron un onfalocele. De estos pacientes, 19 (73%) fueron cerrados de forma primaria y el resto requirió una reducción por etapas con un silo protésico temporal.16
El síndrome de intestino corto, presente en el 25% de los casos, es una causa importante de aumento de la morbilidad nutricional y puede presentarse a pesar de una longitud intestinal normal.4 Una posible alteración absortiva intrínseca del intestino resalta aún más la importancia de una nutrición perioperatoria óptima en estos pacientes y la necesidad de preservar la mayor longitud posible de colon durante la reconstrucción. Si no se utiliza para las heces, el remanente del intestino posterior puede incorporarse durante la reconstrucción del tracto urogenital.17 Aunque existen múltiples vías para el manejo inicial del tracto intestinal, el trabajo de Sawaya et al condujo a un cambio de paradigma en 2009 con la tubularización inicial de la placa cecal y la confección de una colostomía terminal, ya que esto redujo drásticamente la aparición del síndrome de intestino corto y permitió la realización de procedimientos de descenso (pull-through) en el futuro en los candidatos elegibles.20
Se observan otras anomalías asociadas del tracto gastrointestinal en el 46% de los casos e incluyen anomalías de duplicación, gastrosquisis, malrotación, atresia duodenal y segmentos colónicos extrofiados.16,17,21 El ano imperforado también se observa con frecuencia, y una serie informó su presencia en 36 de 37 pacientes.17
Otras anomalías urológicas
Las anomalías del tracto urinario superior son frecuentes y se presentan en el 41–66% de los pacientes.17,18 Es fundamental prestar especial atención a la identificación y comprensión de la anatomía del tracto urinario superior del paciente, ya que la agenesia renal unilateral, el riñón pélvico o la hidronefrosis pueden observarse en hasta el 48% de los casos, con múltiples anomalías renales concomitantes en el 16% de los pacientes.(missing reference) Los riñones en herradura, las anomalías de fusión y las alteraciones ureterales, incluidos el megauréter y la duplicación, se reportan con menor frecuencia. Cabe destacar que las anomalías ureterales pueden variar ampliamente, con posible inserción ectópica del uréter en el conducto deferente en el varón y en el útero, la vagina o las trompas de Falopio en las mujeres.19
Sistema nervioso central y anomalías vertebrales
El disrafismo espinal, que incluye médula anclada, mielomeningocele o lipomielomeningocele, está presente en alguna forma en el 64-100% de los pacientes, y en el 80% se localiza en la región lumbar.2,18 Las anomalías vertebrales distintas del disrafismo incluyen hemivértebras y escoliosis asociada, que se observa en el 40% de los pacientes, aunque la escoliosis no progresa en la mayoría de los casos.15 Debido a esta alta incidencia de defectos espinales y vertebrales, está indicada la evaluación mediante RM de un recién nacido con CE, con participación de neurocirugía según sea necesario.
El compromiso neurológico puede, en última instancia, afectar la función vesical, la continencia urinaria y el movimiento de las extremidades inferiores. El grado en que cada uno puede verse afectado es tan variable como las anomalías espinales y vertebrales. Además, la microdisección de una pelvis infantil con CE en el contexto post mortem reveló un origen y un aporte vascular anómalos de los nervios autónomos.22 Esta imprevisibilidad neurológica se suma al desafío de ayudar a estos pacientes a lograr la continencia. Defectos de la pared abdominal
La ruptura prematura de la membrana cloacal puede resultar en un gran defecto con ausencia completa de la pared abdominal inferior. Si bien la presentación de este defecto es fácilmente evidente, existen sutilezas que requieren una atención particular. Como resultado de la diástasis púbica ensanchada y de cambios en la orientación oblicua típica del canal inguinal, se observan hernias inguinales en hasta el 50% de los pacientes con CE y deben repararse.23
Anomalías que afectan a los órganos reproductivos y a los genitales
En el paciente varón con CE, es típica la separación asimétrica completa del falo en dos mitades, con separación concordante de las mitades escrotales. Los testículos pueden estar situados en sus respectivos hemiescrotos; sin embargo, a menudo están no descendidos y se asocian con hernias inguinales, por lo que requieren corrección quirúrgica. En las mujeres, los cuerpos clitorídeos están de manera similar ampliamente separados.
Las anomalías müllerianas también son comunes en la CE, observándose duplicación uterina en el 95% de los casos.19 Predomina la duplicación parcial, siendo el útero bicorne el subtipo más común. Las anomalías vaginales van desde la agenesia, observada en el 25-50% de los pacientes, hasta la duplicación vaginal, que ocurre en hasta el 65% de los casos.21 En casos de duplicación uterina o de las trompas de Falopio, se debe priorizar la preservación para su posible incorporación a la reconstrucción del tracto urinario inferior.
Anomalías cardiovasculares y pulmonares
Rara vez, la CE se presenta con defectos cardiovasculares o pulmonares que amenazan la vida de forma inmediata. Los informes, aunque poco frecuentes, incluyen una plétora de anomalías mayores, entre ellas duplicación aórtica y duplicación de la vena cava.23 Otros han descrito tetralogía de Fallot, atresia pulmonar, estenosis de la arteria pulmonar y conducto arterioso persistente.24
Dado el impacto asociado de este defecto congénito en el desarrollo de casi todos los sistemas orgánicos, la atención de estos pacientes vulnerables debe proporcionarse en un centro con experiencia en las técnicas quirúrgicas y con un manejo perioperatorio y posoperatorio interdisciplinario.
Evaluación y diagnóstico
Establecimiento del diagnóstico
Considerando la variedad de defectos asociados con la CE, el diagnóstico prenatal ha resultado históricamente difícil, si no esquivo. Diagnosticar con precisión la CE y distinguirla de la CBE es fundamental para asesorar adecuadamente a las familias, especialmente si se considera que los niños con CE tienden a enfrentar más problemas relacionados con la salud a lo largo de su vida. El diagnóstico prenatal permite una planificación que puede optimizar el manejo médico y quirúrgico posnatal. El primer diagnóstico prenatal de CE en 1985 se basó en tres hallazgos clave en la ecografía fetal (fUS): 1. un gran defecto abdominal anterior infraumbilical en la línea media, 2. mielomingocele lumbosacro, y 3. imposibilidad de visualizar la vejiga urinaria.25 Austin y sus colegas refinaron aún más esta lista al delimitar criterios diagnósticos como hallazgos mayores o menores. Los observados en >50% de los casos constituían un criterio mayor e incluían: no visualización de la vejiga (91%), un gran defecto de la pared anterior infraumbilical en la línea media o una estructura quística de la pared anterior (82%), onfalocele (77%) y mielomeningocele (68%). Los criterios menores eran aquellos observados en <50% de los casos e incluían: defectos de las extremidades inferiores (23%), anomalías renales (23%), ascitis (41%), arcos púbicos ensanchados (18%), tórax estrecho (9%), hidrocefalia (9%) y arteria umbilical única (9%).26 A pesar de las grandes mejoras en la fUS desde que se introdujo por primera vez como herramienta diagnóstica, solo un estimado del 15% de los pacientes han sido diagnosticados prenatalmente con la fUS únicamente, ya que los hallazgos pueden identificarse de manera incompleta como onfalocele aislado, CBE u otros defectos de la línea media.
La aparición de la resonancia magnética fetal (fMRI) en el diagnóstico prenatal ha añadido un valioso complemento para la evaluación de la CE. En comparación con la fUS, la fMRI proporciona una visualización anatómica superior cuando no se identifica la vejiga y también puede ayudar a evaluar la presencia o ausencia de un onfalocele, defectos espinales asociados y el sexo, cuando no se identifican fácilmente en la US.27,28
Recientemente, Weiss et al identificaron hallazgos anatómicos clave en fUS y fMRI para evaluar su validez respectiva en el diagnóstico prenatal de CBE y CE. Entre 2001 y 2018 identificaron a 21 pacientes que habían sido sometidos a estudios de imagen prenatales. CBE fue el diagnóstico posnatal en 14 y CE en los restantes. 15 de 21 pacientes tenían tanto fUS como fMRI disponibles para revisión y la mediana de la edad gestacional para la evaluación mediante imagen prenatal fue de 25 semanas. De los 16 fUS con interpretaciones iniciales disponibles, el diagnóstico prenatal original fue correcto en 12 casos, lo que arroja una sensibilidad del 69% para fUS. Los 4 casos de diagnósticos prenatales incorrectos de CE se determinaron posteriormente como CBE. De las 18 fMRI incluidas en el análisis, 16 de 18 diagnósticos concordaron (83% de sensibilidad) y los dos diagnósticos prenatales incorrectos de CE se reclasificaron como CBE. Estos errores diagnósticos se atribuyeron a una gran placa vesical prominente, con asas intestinales posteriormente, simulando un onfalocele con contenido intestinal. Concluyeron que la identificación del punto de inserción del cordón umbilical tanto en fUS como en fMRI era prudente para diferenciar entre CBE y CE dada la presencia de defectos de la pared abdominal.29
Las variantes de la extrofia cloacal plantean desafíos diagnósticos adicionales. Aunque extremadamente rara, también es posible una variante cubierta por piel, como se describió en 5 de 6 pacientes en una pequeña serie de casos.30
Evaluación y manejo posnatal inmediato
Tras el nacimiento, es esencial garantizar que el paciente esté médicamente estable. Se requiere un examen físico exhaustivo para confirmar las diversas anomalías anatómicas que puedan estar presentes (Tabla 1). Dilucidar los defectos congénitos asociados y su gravedad ayuda al manejo médico y quirúrgico. Al igual que en los pacientes con CBE, los segmentos extrofiados del intestino y la vejiga se mantienen húmedos y protegidos con un apósito plástico.31 Una serie completa de estudios por imágenes, incluidos radiografías simples, ecografía y RM, ayuda a determinar la gravedad de los defectos asociados y facilita la participación temprana de servicios de consulta como cirugía ortopédica pediátrica, neurocirugía pediátrica y cirugía general pediátrica. Completar el equipo multidisciplinario con el aporte y el apoyo de trabajo social y de los servicios de nutrición es clave para el cuidado del lactante con CE y su familia.
Asignación de género
Una vez que el lactante se encuentre médicamente estable y se comprenda la extensión completa de la afección, es necesaria una conversación honesta y reflexiva respecto a la asignación de género. En muchos casos, esto puede requerir la consulta con endocrinología pediátrica, psicología infantil o psiquiatría pediátrica. Todas las decisiones sobre la asignación de género deben tomarse únicamente después de realizar un cariotipo, de una discusión exhaustiva y del asesoramiento adecuado a los padres.
Dada la expresión fenotípica típica con amplia separación de los cuerpos cavernosos y del escroto y tamaño diminuto de los cuerpos cavernosos en los niños con CE, los informes iniciales abogaron por la reasignación de género universal de niños 46, XY a mujeres funcionales.32 Esto impulsó los principios tempranos del manejo, que incluían orquiectomía bilateral junto con reconstrucción fálica como clítoris funcional y vaginoplastia, ya sea temprana o diferida.
Los efectos a largo plazo de esta práctica son actualmente tema de intenso debate. Los pacientes que viven más tiempo con esta afección ofrecen una visión de las implicaciones psicosociales de esta práctica y del posible papel del genotipo y del medio hormonal intrauterino. En una cohorte de 29 varones con CE que se sometieron a reasignación de género al femenino, los 29 pacientes mostraron un desplazamiento predominantemente masculino en el desarrollo psicosexual a pesar de no experimentar ningún pico hormonal puberal.33 Otras series, sin embargo, no han demostrado diferencias en el comportamiento ni en problemas psicosociales, aunque se informó un caso de masculinización en un paciente 46XY con conversión de género debido a un testículo ectópico.34,35 Las actitudes actuales favorecen asignar un género concordante con el cariotipo, si es posible. Esto fue respaldado por una encuesta entre urólogos pediátricos en la que dos tercios favorecieron la reconstrucción concordante con el género.36
Manejo quirúrgico y resultados
Manejo inicial
Se recomienda el cierre precoz del onfalocele en el periodo neonatal para
proteger contra una ruptura intempestiva; sin embargo, esto solo se realiza después de abordar las consideraciones neuroquirúrgicas. En el momento del cierre inicial del onfalocele, suele emplearse una de tres vías para la derivación intestinal y el manejo del intestino posterior, incluida la creación de una ileostomía con resección del intestino posterior, la colocación de una ileostomía con fístula mucosa del intestino posterior o la tubularización cecal con creación de una colostomía terminal.
Históricamente, la derivación intestinal inicial se basaba en la ileostomía con resección del intestino posterior; sin embargo, esto acarreaba varias consecuencias no deseadas. En primer lugar, esto inducía universalmente el síndrome de intestino corto y hacía menos probable la reconstrucción posterior del tracto gastrointestinal. En segundo lugar, las ileostomías de alto débito predisponían a los niños a un aumento de las hospitalizaciones por deshidratación recurrente y alteraciones electrolíticas.18,20 La acidosis inducida secundaria al alto débito de la ileostomía afecta negativamente la homeostasis del calcio y modula directamente el eje hormona de crecimiento-IFG-1, atenuando la liberación de la hormona de crecimiento. A largo plazo, esto se asocia con una morbilidad relacionada con el crecimiento en pacientes con CE.37
Debido a esto, ha habido un cambio de paradigma en la derivación intestinal inicial y el manejo del intestino posterior. En su serie de 77 pacientes, Sawaya et al encontraron que la longitud del intestino posterior variaba ampliamente de 2 a >20 cm, con más de la mitad en el rango de 6 a 15 cm. De manera interesante, los pacientes con genotipo XY tenían mayor probabilidad de tener una longitud del intestino posterior inferior a 10 cm en comparación con sus contrapartes XX. En esta serie, solo 10 pacientes se sometieron a resección del intestino posterior y únicamente por viabilidad cuestionable del intestino posterior o por una longitud extremadamente corta. Como resultado, los autores abogaron por la tubularización cecal con creación de colostomía terminal para eliminar el síndrome de intestino corto y facilitar los procedimientos de pull-through intestinal.20 Por lo general, la reconstrucción gastrointestinal se realiza 1 a 2 años después de la derivación fecal inicial; sin embargo, si esta reconstrucción se combina con el cierre vesical, la aproximación del pubis es primordial para una reconstrucción exitosa de la vejiga, la pared abdominal y el intestino. Por lo general, el éxito de estas reconstrucciones depende de la reconstrucción pélvica con osteotomías.17
En su misma serie, Sawaya et al realizaron ocho procedimientos de pull-through intestinal—7 antes de los 5 años de edad, con un rango de 2 a 12 años de edad—y todos los pacientes conservaban un intestino posterior de al menos 10 cm de longitud.20 Otros factores a considerar al decidir si realizar un pull-through intestinal incluyen la capacidad del paciente para formar heces sólidas y la evidencia de musculatura esfinteriana anal que responde a la estimulación cuando se examina bajo anestesia. En los niños que no son candidatos para el pull-through intestinal, un estoma fecal permanente es la opción más duradera para el manejo a largo plazo. Cabe señalar que, si el remanente del intestino posterior no se incorpora al tránsito fecal, debe preservarse para una posterior ampliación vesical o reconstrucción vaginal.17
Si en la fase inicial del cierre de la onfalocele se determina que no puede lograrse de manera segura y exitosa el cierre simultáneo de la vejiga y la pared abdominal, entonces las mitades vesicales deben aproximarse en la línea media, convirtiendo el defecto en una CBE.17 Esto permite la distensión abdominal y el agrandamiento de la placa vesical para un cierre diferido posterior. Por último, el intestino posterior se deja como fístula mucosa en este momento si no se incorpora en la reconstrucción intestinal inicial.
Reconstrucción urinaria
Reparación moderna en etapas
La reconstrucción urinaria de CE es paralela a la de CBE. La aproximación posterior de las hemivejigas convierte el defecto de CE en CBE e implica la disección de las caras laterales de las mitades vesicales desde la pared abdominal y el cierre en la línea media.38 Restaurar la anatomía mediante la colocación de la vejiga y la uretra posterior en profundidad dentro de la pelvis sigue siendo un factor crucial para una reconstrucción quirúrgica exitosa. La aproximación de la pelvis ensanchada ayuda a este objetivo, permitiendo la reconstrucción de la pared abdominal y del tracto urinario; sin embargo, esto suele requerir osteotomías pélvicas y fijación, como se describirá más adelante en este capítulo.
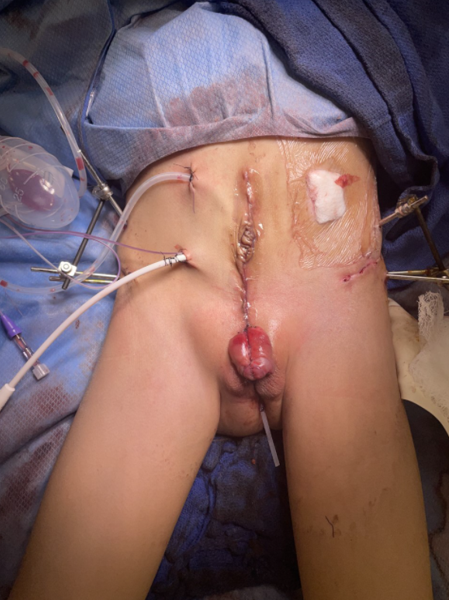
Figura 6 Reparación moderna por etapas de la extrofia. Fotografía posoperatoria del paciente visto en la fotografía preoperatoria en Figura 3. Nótese la colocación del pin de osteotomía antes de la aplicación del dispositivo de fijación externa.
El gran defecto de la pared abdominal que se observa en la CE supone un desafío significativo para la reconstrucción de la pared abdominal sin tensión (Figura 7). No tener en cuenta la tensión en el momento del cierre puede afectar las tasas de cierre exitoso y de formación de fístulas. Los materiales bioprotésicos como Alloderm (Allergan, Branchburg, NJ) han mostrado gran promesa para puentear el defecto debido a la falta de tejido de la pared abdominal, a la vez que proporcionan un cierre sin tensión.39 Además, Alloderm se ha utilizado como adyuvante para disminuir la fistulización penopúbica mediante la cobertura de la sutura interpúbica en el momento de la reconstrucción del tracto urinario.40
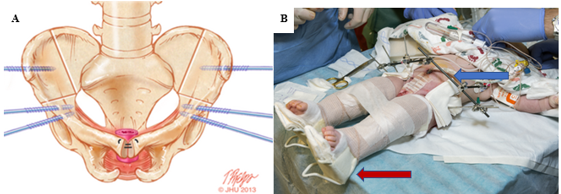
Figura 7 A, sitios de osteotomías combinadas bilaterales: innominada anterior e ilíaca vertical, marcados en el hueso, con los puntos ideales de inserción de los pernos. B, tracción de Buck modificada (flecha roja) y fijación externa (flecha azul) a la derecha.
Para garantizar la congruencia con el género asignado, la reconstrucción de los genitales externos se realiza tan pronto como sea posible en el periodo posnatal inmediato. En los varones genotípicos, esto garantizará que sean criados de manera acorde con su fenotipo. Aunque los efectos psicosociales y psicosexuales a largo plazo en los niños que se someten a reasignación de género son actualmente objeto de gran interés, como se comentó anteriormente, los estudios histológicos de los testículos en sujetos masculinos que se sometieron a reasignación de género confirman una histología normal, incluso pese a la criptorquidia.41 Dado que el tejido peniano presente suele ser escaso y asimétrico, la reconstrucción fálica sigue siendo un desafío, con resultados mixtos.
La faloplastia ha surgido como una opción reconstructiva exitosa para el reemplazo peniano, aunque por lo general se pospone hasta la adolescencia tardía (Figura 8).42 Sin embargo, en los casos en que la escasez de tejido exige una reasignación de masculino a femenino, la reconstrucción genital inicial debe unir las mitades fálicas en la línea media para formar un clítoris. Cuando el tejido fálico es adecuado, sin embargo, la reparación del epispadias se realiza al año de edad, a menudo utilizando la bien descrita reparación de Cantwell-Ransley.43 En personas genéticamente femeninas, la reconstrucción vaginal se realiza de forma temprana, y en varones reasignados de género, la reconstrucción diferida de una neovagina es apropiada, requiriéndose la dilatación a largo plazo de la neovagina.44 Debido a la uretra más corta en las personas genéticamente femeninas, la reparación del epispadias aislado suele combinarse con reconstrucción del cuello vesical de Young-Dees-Leadbetter (BNR), monoplastia y clitoroplastia.
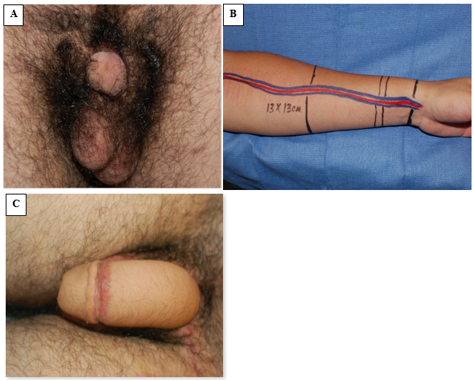
Figura 8 Faloplastia con colgajo libre de antebrazo radial. A, fotografía preoperatoria con escaso tejido peneano. B, preparación intraoperatoria del tejido del colgajo de antebrazo y de la vasculatura pediculada asociada. C, fotografía posoperatoria que demuestra el resultado de la faloplastia.
La importancia de la osteotomía
La osteotomía ha surgido como un factor crítico para el cierre exitoso en la mayoría de los niños con CBE y está indicada en todos los niños con CE en el momento del cierre vesical.17,44 La osteotomía corrige la amplia diástasis púbica y permite la colocación ortotópica de la vejiga y de la uretra posterior lo más profundamente posible dentro de la pelvis, al mismo tiempo que facilita la reconstrucción vesical y de la pared abdominal con tensión mínima. Esta disminución de la tensión se correlaciona con tasas reducidas de dehiscencia y de hernias ventrales posoperatorias. Además, se ha observado que la osteotomía reduce la probabilidad de complicaciones significativas del 89% en pacientes cerrados sin osteotomía al 17% en niños cerrados con osteotomía.45 Esto se confirmó en una gran serie de 80 pacientes con CE que informó un 91% de reparaciones exitosas de la extrofia con osteotomía realizada en el momento del cierre.46 Si bien la osteotomía ha ayudado a lograr cierres exitosos, no se ha demostrado que afecte la continencia definitiva en los pacientes con CE.
Las osteotomías combinadas bilaterales innominadas anteriores y ilíacas verticales son el método preferido en nuestra institución y se coordinan con el cierre vesical en un enfoque denominado Dual-Staged Pathway (DSP).47 Este enfoque de osteotomía en posición supina brinda el beneficio adicional de no necesitar reposicionar al paciente antes del cierre vesical y de la pared abdominal (Figura 7). Al evitarse un abordaje posterior, también se minimizan los riesgos de dañar cualquier reparación espinal. Además, las osteotomías pélvicas permiten una reducción pélvica gradual con fijación externa entre 2 y 3 semanas antes del cierre vesical y de la pared abdominal. Esto se utiliza con frecuencia en casos de diástasis púbica extremadamente amplia (> 10 cm) y se empleó con éxito en un paciente con una diástasis púbica de 16 cm.47 En nuestra institución, la fijación externa y la tracción de Buck modificada se mantienen durante 6 a 8 semanas para asegurar una cicatrización adecuada. En nuestra experiencia, esta técnica de inmovilización proporciona resultados excepcionales con una tasa de fracaso del 3.8% en cierres primarios, en comparación con el 65.7% para la inmovilización con un yeso en espica.48
Reconstrucción en una sola etapa
Otro enfoque de reparación en el paciente con CE es la reparación en un solo paso, o reparación primaria completa de la extrofia (CPRE), popularizada por Grady y Mitchell.49 A diferencia del enfoque por etapas de reconstrucción abdominal y vesical, seguido de la reparación del epispadias y un procedimiento de continencia, la reparación en una sola etapa combina aspectos de cada etapa en una única reparación. El procedimiento es similar al descrito para CBE; sin embargo, tiene en cuenta el momento del cierre con respecto al onfalocele, enfatizando retrasar el cierre en caso de un onfalocele grande u otras circunstancias médicas atenuantes. Otros han concluido que, si bien la reparación en una sola etapa es aplicable a un grupo seleccionado de pacientes, otros factores que pueden impedir su uso son una placa vesical pequeña, diástasis púbica >6 cm u otras condiciones médicas como la disrafia espinal, que, incluso en ausencia de extrofia, pueden requerir múltiples procedimientos quirúrgicos.50 En una serie limitada de 6 pacientes, Lee et al informaron un cierre exitoso con reparación primaria completa. No obstante, señalaron el desarrollo de un tabique vesical en la línea media con hidronefrosis asociada en dos niños. Además, un niño miccionó de forma espontánea y otro se sometió a cistoplastia de aumento (AC).51
Los defensores de esta técnica sostienen que puede disminuir los costos, la morbilidad y el traumatismo asociados con múltiples operaciones, e incluso estimular el crecimiento vesical precoz. En este caso, la reparación del epispadias se realiza mediante desensamblaje peniano, en el que la placa uretral se diseca por completo de los cuerpos cavernosos. Esto permite el cierre uretral y sitúa el cuello vesical posteriormente dentro de la pelvis.52 Los defensores de la CPRE atribuyen la disminución del número de operaciones necesarias para lograr la continencia al aumento de la resistencia del tracto de salida vesical a una edad temprana. Sin embargo, muchos pacientes aún requieren BNR y, notablemente, solo el 20-56% de los pacientes con CPRE que permanecen incontinentes logran la continencia tras una BNR adicional.53,54
Complicaciones
Tanto las reparaciones en un solo tiempo como las por etapas son técnicas de reconstrucción aceptables en la CE. Aunque cada enfoque tiene sus méritos, los resultados comunicados sitúan la tasa de fracaso de una reconstrucción en un solo tiempo en 48% en comparación con 15% para una reparación por etapas. La dehiscencia de la herida, el prolapso vesical, la obstrucción del tracto de salida vesical y la formación de una fístula vesicocutánea deben considerarse cierres fallidos y se han comunicado en ambas técnicas, en un solo tiempo y por etapas.
Con cualquiera de los abordajes quirúrgicos, el cierre vesical primario exitoso es el objetivo principal. Un cierre fallido es devastador para el paciente y la familia, y las repercusiones son significativas. Los pacientes que experimentan un cierre primario fallido en CE son sometidos a más operaciones y a una mayor exposición a anestesia general. Aunque un cierre vesical fallido en CBE impacta negativamente la continencia urinaria definitiva, este impacto específico sobre la continencia es menos pronunciado en los pacientes con CE, ya que la mayoría de estos pacientes requieren procedimientos adicionales de continencia independientemente del resultado primario.55,56 Las repercusiones se extienden más allá de las operaciones adicionales e incluyen morbilidad financiera. Goldstein et al realizaron un análisis de costos en pacientes con CE y encontraron que, con una reparación primaria exitosa de CE, cuesta aproximadamente $196,000 alcanzar la continencia, mientras que el costo para lograr la continencia aumenta a $407,000 después de un cierre vesical primario fallido.57
Una serie amplia que evaluó los predictores de un cierre primario exitoso en la extrofia informó que un cierre CE por etapas, junto con el uso de osteotomía pélvica, fueron predictores independientes significativos de un cierre exitoso. Además, después del cierre vesical, la inmovilización posoperatoria fue crucial para el éxito.58 Por este motivo, recomendamos el uso de fijación externa combinada con tracción, como es la norma en nuestra institución. Otros métodos, como los yesos espica, pueden no proporcionar una inmovilización adecuada y, por lo tanto, pueden conducir a un mayor fracaso de la reconstrucción pélvica. Las complicaciones de la osteotomía son poco frecuentes y a menudo autolimitadas, pero incluyen un mayor riesgo de parálisis nerviosas y musculares transitorias (que suelen resolverse dentro de las 12 semanas posteriores a la cirugía), retardo de consolidación ilíaca y infección o inflamación superficial del sitio de los pines.59 En la experiencia de la institución de los autores, aunque la osteotomía pélvica con inmovilización posoperatoria puede prolongar la estancia hospitalaria, la drástica reducción en la tasa de fracaso se considera que vale la pena por los pacientes y los proveedores de atención.
Evidencia emergente sugiere que el momento del cierre inicial afecta las tasas de éxito. En 2014, Shah y colegas reportaron resultados en 60 pacientes con CE y encontraron que un aumento de 1 cm en la diástasis previa al cierre resultó en un incremento de 2,64 en las probabilidades de fracaso del cierre inicial. Realizar posteriormente una osteotomía y retrasar el cierre, permitiendo que el fijador externo disminuyera la diástasis, incrementó el éxito del cierre. Curiosamente, el 77% de los pacientes con un cierre fallido fueron cerrados dentro de la primera semana de vida en comparación con el 26% de los pacientes que se sometieron a cierre por etapas. Además, solo el 31% de los pacientes del grupo de cierre fallido se sometieron a osteotomía durante el cierre inicial en comparación con el 82% de los pacientes con cierre por etapas. Otras estrategias protectoras contra el fracaso incluyen retrasar el cierre vesical inicial hasta después del cierre del onfalocele.60 Por último, el 92% de los pacientes con un recierre exitoso se sometieron a osteotomía.47 Estos resultados, que demuestran un mayor éxito en la reparación primaria diferida, se apoyaron en otra serie que demostró un mayor porcentaje de fracasos en el cierre vesical primario en pacientes cerrados dentro de los primeros 30 días de vida en comparación con aquellos realizados después de los 30 días de edad (47,7% frente a 19,3%).55
Para lograr la continencia tras un cierre primario fallido, debe realizarse un cierre vesical secundario. De forma similar a los hallazgos reportados por Shah et al., Davis y sus colegas encontraron que la edad a la que se realiza el cierre vesical secundario es predictiva del éxito global. Informaron que los pacientes con un cierre vesical secundario exitoso tenían más de un año más de edad que aquellos en quienes fracasó el cierre vesical secundario (edad mediana 104 semanas vs 38 semanas).56 El cierre secundario exitoso presentó un retraso mediano de 154 semanas, en comparación con un retraso mediano de 30 semanas en los cierres secundarios fallidos. El fracaso de la reconstrucción urinaria en niños con extrofia vesical es significativo, ya que puede conducir a un crecimiento vesical atenuado y a la pérdida de la plantilla vesical, lo que puede hacer que lograr la continencia sea cada vez más difícil, si no imposible.
Lograr la continencia
Debido a la constelación frecuente de posibles anomalías—placas vesicales pequeñas, vejigas con baja complacencia, cuello vesical ampliamente patente (si es que está presente) y anomalías espinales que contribuyen a intestino y vejiga neurogénicos—la continencia miccional es poco probable. No obstante, la continencia sigue siendo el objetivo último en la reconstrucción de la extrofia para permitir que estos niños lleven vidas más típicas. Con este fin, a menudo se requieren procedimientos adicionales, como confirmó Maruf et al en una gran serie que informó que los pacientes requieren una mediana de 2 (rango 1–4) procedimientos de continencia urinaria para lograr un intervalo seco mayor de 3 horas.61
En una amplia serie de la institución de los autores que evaluó los desenlaces en 80 pacientes con complejo OEIS, se disponía de datos sobre reconstrucción urinaria para 73 pacientes. De los 40 pacientes secos, 38 conservaron su vejiga nativa, mientras que 2 tenían neovejigas de intestino delgado (bolsa de Koch). De los 38 pacientes con vejiga nativa, 30 (79%) recibieron un canal cateterizable continente (derivación urinaria continente, CUD) con apéndice (n =3) o con segmento intestinal (n = 27), y 32 (84%) también tuvieron cistoplastia de aumento (AC) antes de alcanzar la sequedad. Solo 9 pacientes (24%) mantuvieron una uretra permeable y continente.46 En un conjunto de estudios que evaluaron la continencia urinaria en CE, se requirió AC en hasta el 50% de los pacientes y una tasa global de sequedad del 72%.62
Estos procedimientos adicionales para la continencia, AC y CUD, presentan complicaciones particulares, que incluyen sobreproducción de moco, cálculos vesicales, colonización bacteriana crónica y pólipos epiteliales. Otros problemas a largo plazo se derivan de la AC e incluyen acidosis metabólica crónica y, con menor frecuencia, carcinoma.63,64 Con el tiempo, el estoma creado para CUD puede estenosarse, prolapsar, necrosarse o presentar fugas, lo que requiere revisión.64
En pacientes con sexo genético femenino, puede lograrse continencia exitosa tras BNR de Young-Dees-Leadbetter, aunque la mayoría de las pacientes requieren cateterismo intermitente limpio. Las técnicas utilizadas tanto en varones como en mujeres para aumentar la capacidad vesical se describieron previamente, pero incluyen incorporar el segmento del intestino posterior (cuando está disponible), el intestino delgado y el grueso y, con menor frecuencia, el estómago.38 Lograr la continencia es mucho más desafiante en la paciente reasignada de varón a mujer y un estoma continente puede ser la estrategia más ventajosa a largo plazo.17 Independientemente de las técnicas empleadas, en la experiencia de nuestra institución, los niños son continentes de orina a una edad mediana de 11 años.61 En una serie grande que evaluó la continencia en pacientes con extrofia cloacal, el 50% (35 de 61) de los niños alcanzaron la continencia, con 30 de 35 realizando cateterismo intermitente a través de un estoma continente y el resto miccionando o cateterizándose por uretra.65
Seguimiento recomendado
Los pacientes con extrofia vesical, sus familias y los equipos médicos y quirúrgicos tratantes desarrollan un vínculo especial que es único en la medicina. Tras la reconstrucción del tracto urinario y de la pared abdominal, y una posible reparación posterior del epispadias, el enfoque del manejo se desplaza hacia la protección de las vías urinarias superiores. Con este fin, en la institución del autor, vigilamos cualquier evidencia de reflujo mediante cistografías anuales y cistoscopia, al tiempo que seguimos el crecimiento vesical. En los niños en edad escolar (al menos de 5 a 7 años) evaluamos la capacidad vesical, los estudios urodinámicos y la capacidad del niño para participar en su propio cuidado, mediante la valoración de la destreza y la madurez, antes de la reconstrucción del cuello vesical.31
Conclusiones
CE es una malformación congénita multisistémica devastadora que anteriormente predisponía a los niños a una morbilidad significativa e incluso mortalidad. Sin embargo, grandes avances en el manejo quirúrgico y médico de estos niños les han permitido llevar vidas casi normales hasta la edad adulta. El éxito en el manejo contemporáneo se basa en el enfoque multidisciplinario para superar los desafíos que enfrentan estos pacientes. A medida que estos pacientes continúen viviendo vidas saludables bien entrada la edad adulta, seguiremos aprendiendo más en cuanto a la calidad de vida y las formas en que puede optimizarse la función global del paciente.
Puntos clave
- Un diagnóstico perinatal preciso y exhaustivo es crucial para organizar una atención multidisciplinaria adecuada—fUS y fMRI son modalidades útiles en este empeño.
- El manejo postnatal inicial debe garantizar que el paciente esté médicamente estable. Una vez estable, se debe realizar un examen físico exhaustivo y utilizar las técnicas de imagen adecuadas para asegurar la identificación completa de todas las anomalías asociadas.
- La reconstrucción abdominal y del tracto urinario solo puede llevarse a cabo después de la evaluación neuroquirúrgica de la posible afectación de la columna o del SNC y su autorización.
- Cualquier remanente de intestino posterior debe preservarse a toda costa en estos niños, ya que puede utilizarse en la reconstrucción gastrointestinal o genitourinaria.
- La separación de la vejiga del tracto gastrointestinal con una derivación fecal concomitante es fundamental antes de la reconstrucción futura. La colostomía terminal se ha consolidado como la forma de derivación fecal de menor morbilidad.
- Un seguimiento estrecho tras la reconstrucción en estos pacientes es esencial para asegurar la preservación de la función del tracto urinario superior.
Direcciones futuras
Las investigaciones actuales buscan mejorar el asesoramiento y disminuir la morbilidad asociada a la cirugía mediante el desarrollo de mejores herramientas predictivas para los pacientes y sus familias. Un mejor asesoramiento puede lograrse mediante herramientas, como aquellas que predicen la capacidad vesical y el crecimiento (Sholklapper et al 2022, enviado), lo cual puede tener implicaciones en cirugías futuras y en la capacidad de lograr la continencia.66 Además, la investigación en ciencias básicas que evalúa las diferencias de ultraestructura entre las vejigas con extrofia y los controles normales apareados por edad puede ofrecer una imagen más clara de las curvas diferenciales de crecimiento vesical para estos pacientes. El uso de técnicas de imagen para evaluar la anatomía del suelo pélvico como herramienta de planificación quirúrgica proporciona información adicional para la toma de decisiones intraoperatorias y se espera que el empleo de estas técnicas se expanda. A largo plazo, abordar el creciente campo de la urología traslacional y definir quién atiende a estos niños cuando ingresan a la edad adulta ayudará a optimizar la atención de estos pacientes. Por último, la expansión de la telemedicina para la consulta entre centros médicos puede mejorar la comunicación respecto de esta afección infrecuente y mejorar el bienestar de los pacientes y sus familias al reducir la carga relacionada con la accesibilidad, los desplazamientos y los costos.
Lecturas recomendadas
- Gearhart JP, Carlo HN. Exstrophy-epispadias complex. 12th ed., Philadelphia, PA: Elsevier; 2020, DOI: 10.1201/9780429194993-25.
- Grady RW, Mitchell ME. Complete Primary Repair Of Exstrophy. J Urol 1999; 65 (1): 1415–1420. DOI: 10.1097/00005392-199910000-00071.
- Davis R, Stewart D, Maruf M, Lau H, Gearhart JP. Complex abdominal wall reconstruction combined with bladder closure in OEIS complex. J Pediatr Surg 2019; 54 (11): 2408–2412. DOI: 10.1016/j.jpedsurg.2019.03.022.
- Tourchi A, Inouye BM, Di Carlo HN, Young E, Ko J, Gearhart JP. New advances in the pathophysiologic and radiologic basis of the exstrophy spectrum. Journal of Pediatric Urology 2014; 10 (2): 212–218.
- Kasprenski M, Michaud J, Yang Z, Maruf M, Benz K, Jayman J, et al.. Urothelial differences in the exstrophy-epispadias complex: potential implications for management. The Journal of Urology 2021; 205 (5): 1460–1465.
- Di Carlo HN, Maruf M, Massanyi EZ, Shah B, Tekes A, Gearhart JP. 3-dimensional magnetic resonance imaging guided pelvic floor dissection for bladder exstrophy: a single arm trial. The Journal of Urology 2019; 202 (2): 406–412.
Referencias
- Muecke EC. The Role of the Cloacae Membrane in Exstrophy: The First Successful Experimental Study. J Urol 1069; 92 (6): 659–668. DOI: 10.1016/s0022-5347(17)64028-x.
- Woo LL, Thomas JC, Brock JW. Cloacal exstrophy: A comprehensive review of an uncommon problem. J Pediatr Urol 2010; 6 (2): 102–111. DOI: 10.1016/j.jpurol.2009.09.011.
- Moore KL. Organogenetic Period: The Fourth to Eighth Weeks. In: Moore KL, Persaud TVN, editors. The Developing Human: Clinically Oriented Embryology. 7th ed. Philadelphia, PA: Saunders; 2003.
- Howell C, Caldamone A, Snyder H, Ziegler M, Duckett J. Optimal management of cloacal exstrophy. J Pediatr Surg 1983; 18 (4): 365–369. DOI: 10.1016/s0022-3468(83)80182-1.
- Cervellione RM, Mantovani A, Gearhart J, Bogaert G, Gobet R, Caione P, et al.. Prospective study on the incidence of bladder/cloacal exstrophy and epispadias in Europe. J Pediatr Urol 2015; 11 (6): 337.e1–337.e6. DOI: 10.1016/j.jpurol.2015.03.023.
- Shapiro E, Lepor H, Jeffs RD. The Inheritance of the Exstrophy-Epispadias Complex. J Urol 1984; 132 (2): 308–310. DOI: 10.1016/s0022-5347(17)49605-4.
- Ives E, Coffey R, Carter CO. A family study of bladder exstrophy. J Med Genet 1980; 17 (2): 139–141. DOI: 10.1136/jmg.17.2.139.
- Birth Defects Monitoring Systems IC for. Epidemiology of bladder exstrophy and epispadias: A communication from the international clearinghouse for birth defects monitoring systems. Teratology 1987; 36 (2): 221–227. DOI: 10.1002/tera.1420360210.
- Botto LD, Feldkamp ML, Amar E, Carey JC, Castilla EE, Clementi M, et al.. Acardia: Epidemiologic findings and literature review from the International Clearinghouse for Birth Defects Surveillance and Research. Am J Med Genet C Semin Med Genet 2011; 157 (4): 262–273. DOI: 10.1002/ajmg.c.30318.
- Boyadjiev SA, Dodson JL, Radford CL, Ashrafi GH, Beaty TH, Mathews RI, et al.. Clinical and molecular characterization of the bladder exstrophy-epispadias complex: analysis of 232 families. BJU Int 2004; 94 (9): 1337–1343. DOI: 10.1111/j.1464-410x.2004.05170.x.
- WOOD HADLEYM, TROCK BRUCEJ, GEARHART JOHNP. In Vitro Fertilization and the Cloacal-Bladder Exstrophy-Epispadias Complex: Is there an Association? J Urol 2003; 169 (4): 1512–1515. DOI: 10.1097/01.ju.0000054984.76384.66.
- Reutter H, Boyadjiev SA, Gambhir L, Ebert A-K, Rösch WH, Stein R, et al.. Phenotype Severity in the Bladder Exstrophy-Epispadias Complex: Analysis of Genetic and Nongenetic Contributing Factors in 441 Families from North America and Europe. J Pediatr 2011; 159 (5): 825–831.e1. DOI: 10.1016/j.jpeds.2011.04.042.
- Sponseller PD, Bisson LJ, Gearhart JP, Jeffs RD, Magid D, Fishman E. The anatomy of the pelvis in the exstrophy complex. J Bone Joint Surg Am 1995; 77 (2): 177–189. DOI: 10.2106/00004623-199502000-00003.
- Stec AA, Pannu HK, Tadros YE, Sponseller PD, Fishman EK, Gearhart JP. Pelvic Floor Anatomy In Classic Bladder Exstrophy Using 3-dimensional Computerized Tomography: J Urol 2001; 166 (4): 1444–1449. DOI: 10.1097/00005392-200110000-00066.
- Greene WB, Dias LS, Lindseth RE, Torch MA. Musculoskeletal problems in association with cloacal exstrophy. J Bone Joint Surg Am 1991; 73 (4): 551–560. DOI: 10.2106/00004623-199173040-00012.
- Davidoff AM, Hebra A, Balmer D, Templeton JM, Schnaufer L. Management of the gastrointestinal tract and nutrition in patients with cloacal exstrophy. J Pediatr Surg 1996; 31 (6): 771–773. DOI: 10.1016/s0022-3468(96)90129-3.
- Mathews R, Jeffs RD, Reiner WG, Docimo SG, Gearhart JP. Cloacal Exstrophy-improving The Quality Of Life. J Urol 1998; 6 (2): 2452–2456. DOI: 10.1097/00005392-199812020-00017.
- McHoney M, Ransley PG, Duffy P, Wilcox DT, Spitz L. Cloacal exstrophy: Morbidity associated with abnormalities of the gastrointestinal tract and spine. J Pediatr Surg 2004; 39 (8): 1209–1213. DOI: 10.1016/j.jpedsurg.2004.04.019.
- Sugar EC, Firlit CF. Management of cloacal exstrophy. Urology 2004; 32 (4): 320–322. DOI: 10.1016/0090-4295(88)90234-8.
- Sawaya D, Goldstein S, Seetharamaiah R, Suson K, Nabaweesi R, Colombani P, et al.. Gastrointestinal ramifications of the cloacal exstrophy complex: a 44-year experience. J Pediatr Surg 2010; 45 (1): 171–176. DOI: 10.1016/j.jpedsurg.2009.10.030.
- Hurwitz RS, Manzoni GAM, Ransley PG, Stephens FD. Cloacal Exstrophy: A Report of 34 Cases. J Urol 1987; 138 (4 Part 2): 1060–1064. DOI: 10.1016/s0022-5347(17)43502-6.
- Phillips TM, Salmasi AH, Stec A, Novak T, Gearhart JP, Mathew J. Re: Urological Outcomes in the Omphalocele Exstrophy Imperforate Anus Spinal Defects (OEIS) Complex: Experience with 80 Patients. J Urol 2013; 191 (4): 1118–1119. DOI: 10.1016/j.juro.2014.01.044.
- Schlegel PN, Gearhart JP. Neuroanatomy of the Pelvis in an Infant with Cloacal Exstrophy: A Detailed Microdissection with Histology. J Urol 1989; 141 (3 Part 1): 583–585. DOI: 10.1016/s0022-5347(17)40901-3.
- Connolly JA, Peppas DS, Jeffs RD, Gearhart JP. Prevalence and Repair of Inguinal Hernias in Children with Bladder Exstrophy. J Urol 1995; 154 (5): 1900–1901. DOI: 10.1097/00005392-199511000-00093.
- Sadula SR, Kanhere SV, Phadke VD. Exstrophy of Cloaca Sequence (Oeis Complex) With Multiple Cardiac Malformations. Indian Journal of Case Reports 2019; 05 (04): 317–319. DOI: 10.32677/ijcr.2019.v05.i04.006.
- Meizner I, Bar-Ziv J. In utero prenatal ultrasonic diagnosis of a rare case of cloacal exstrophy. J Clin Ultrasound 1985; 13 (7): 500–502. DOI: 10.1002/jcu.1870130714.
- Austin PF, Homsy YL, Gearhart JP, Porter K, Guidi C, Madsen K, et al.. The Prenatal Diagnosis Of Cloacal Exstrophy. J Urol 1998; 160 (3.2): 1179–1181. DOI: 10.1097/00005392-199809020-00061.
- Calvo-Garcia MA, Kline-Fath BM, Rubio EI, Merrow AC, Guimaraes CV, Lim F-Y. Fetal MRI of cloacal exstrophy. Pediatr Radiol 2013; 43 (5): 593–604. DOI: 10.1007/s00247-012-2571-3.
- Goto S, Suzumori N, Obayashi S, Mizutani E, Hayashi Y, Sugiura-Ogasawara M. Prenatal findings of omphalocele-exstrophy of the bladder-imperforate anus-spinal defects (OEIS) complex. Congenit Anom (Kyoto) 2012; 52 (3): 179–181. DOI: 10.1111/j.1741-4520.2011.00342.x.
- Weiss DA, Oliver ER, Borer JG, Kryger JV, Roth EB, Groth TW, et al.. Key anatomic findings on fetal ultrasound and MRI in the prenatal diagnosis of bladder and cloacal exstrophy. J Pediatr Urol 2020; 16 (5): 665–671. DOI: 10.1016/j.jpurol.2020.07.024.
- Lowentritt BH, Van Zijl PS, Frimberger D, Baird A, Lakshmanan Y, Gearhart JP. Variants Of The Exstrophy Complex: A Single Institution Experience. J Urol 2005; 173 (5): 1732–1737. DOI: 10.1097/01.ju.0000154353.03056.5c.
- Gearhart JP, Jeffs RD. The bladder exstrophy-epispadias complex. 3rd ed., Philadelphia, PA: Saunders; 1998, DOI: 10.1007/978-3-319-44182-5_13.
- Tank ES, Lindenauer SM. Principles of management of exstrophy of the cloaca. Am J Surg 1970; 119 (1): 95–98. DOI: 10.1016/0002-9610(70)90018-8.
- Reiner WG, Gearhart JP. Discordant Sexual Identity in Some Genetic Males with Cloacal Exstrophy Assigned to Female Sex at Birth. N Engl J Med 2004; 350 (4): 333–341. DOI: 10.1056/nejmoa022236.
- Baker Towell DM, Towell AD. A Preliminary Investigation Into Quality of Life, Psychological Distress and Social Competence in Children With Cloacal Exstrophy. J Urol 2003; 169 (5): 1850–1853. DOI: 10.1097/01.ju.0000062480.01456.34.
- Mirheydar H, Evason K, Coakley F, Baskin LS, DiSandro M. 46, XY female with cloacal exstrophy and masculinization at puberty. J Pediatr Urol 2009; 5 (5): 408–411. DOI: 10.1016/j.jpurol.2009.03.013.
- Diamond DA, Burns JP, Mitchell C, Lamb K, Kartashov AI, Retik AB. Sex assignment for newborns with ambiguous genitalia and exposure to fetal testosterone: Attitudes and practices of pediatric urologists. J Pediatr 2006; 148 (4): 445–449. DOI: 10.1016/j.jpeds.2005.10.043.
- Fullerton BS, Sparks EA, Hall AM, Chan Y-M, Duggan C, Lund DP, et al.. Growth morbidity in patients with cloacal exstrophy: a 42-year experience. J Pediatr Surg 2016; 51 (6): 1017–1021. DOI: 10.1016/j.jpedsurg.2016.02.075.
- Gearhart JP, Jeffs RD. Techniques to Create Urinary Continence in the Cloacal Exstrophy Patient. J Urol 1991; 146 (2 Part 2): 616–618. DOI: 10.1016/s0022-5347(17)37871-0.
- Davis R, Stewart D, Maruf M, Lau H, Gearhart JP. Complex abdominal wall reconstruction combined with bladder closure in OEIS complex. J Pediatr Surg 2019; 54 (11): 2408–2412. DOI: 10.1016/j.jpedsurg.2019.03.022.
- Henderson CG, North AC, Gearhart JP. The use of alloderm as an adjunct in the closure of the bladder – Cloacal exstrophy complex. J Pediatr Urol 2011; 7 (1): 44–47. DOI: 10.1016/j.jpurol.2010.02.209.
- Mathews, Perlman, Marsh, Gearhart. Gonadal morphology in cloacal exstrophy: implications in gender assignment. BJU Int 1999; 84 (1): 99–100. DOI: 10.1046/j.1464-410x.1999.00148.x.
- Lumen N, Monstrey S, Selvaggi G, Ceulemans P, De Cuypere G, Van Laecke E, et al.. Phalloplasty: A Valuable Treatment for Males with Penile Insufficiency. Urology 2008; 71 (2): 272–276. DOI: 10.1016/j.urology.2007.08.066.
- Gearhart JP, Leonard MP, Burgers JK, Jeffs RD. The Cantwell-Ransley Technique for Repair of Epispadias. J Urol 1992; 148 (3 Part 1): 851–854. DOI: 10.1016/s0022-5347(17)36742-3.
- Gearhart JP, Carlo HN. Exstrophy-epispadias complex. 12th ed., Philadelphia, PA: Elsevier; 2020, DOI: 10.1201/9780429194993-25.
- Ben-Chaim J, Peppas DS, Sponseller PD, Jeffs RD, Gearhart JP. Applications of Osteotomy in the Cloacal Exstrophy Patient. J Urol 1995; 154 (2.2): 865–867. DOI: 10.1097/00005392-199508000-00146.
- Mathews R, Gearhart JP, Bhatnagar R, Sponseller P. Staged Pelvic Closure of Extreme Pubic Diastasis in the Exstrophy-Epispadias Complex. J Urol 2006; 176 (5): 2196–2198. DOI: 10.1016/j.juro.2006.07.058.
- Benz KS, Jayman J, Maruf M, Baumgartner T, Kasprenski MC, Friedlander DA, et al.. Pelvic and lower extremity immobilization for cloacal exstrophy bladder and abdominal closure in neonates and older children. J Pediatr Surg 2018; 53 (11): 2160–2163. DOI: 10.1016/j.jpedsurg.2017.11.066.
- Grady RW, Mitchell ME. Complete Primary Repair Of Exstrophy. J Urol 1999; 65 (1): 1415–1420. DOI: 10.1097/00005392-199910000-00071.
- Thomas JC, DeMarco RT, Pope JC, Adams MC, Brock JW. First Stage Approximation of the Exstrophic Bladder in Patients With Cloacal Exstrophy–Should This be the Initial Surgical Approach in all Patients? J Urol 2007; 178 (4s): 1632–1636. DOI: 10.1016/j.juro.2007.03.164.
- Lee RS, Grady R, Joyner B, Casale P, Mitchell M. Can a Complete Primary Repair Approach be Applied to Cloacal Exstrophy? J Urol 2006; 176 (6): 2643–2648. DOI: 10.1016/j.juro.2006.08.052.
- Mitchell ME, Bagli DJ. Complete Penile Disassembly for Epispadias Repair. J Urol 1996; 155 (1): 300–304. DOI: 10.1097/00005392-199601000-00128.
- Gargollo P, Hendren WH, Diamond DA, Pennison M, Grant R, Rosoklija I, et al.. Bladder Neck Reconstruction is Often Necessary After Complete Primary Repair of Exstrophy. J Urol 2011; 185 (6s): 2563–2571. DOI: 10.1016/j.juro.2011.01.024.
- Schaeffer AJ, Stec AA, Purves JT, Cervellione RM, Nelson CP, Gearhart JP. Complete Primary Repair of Bladder Exstrophy: A Single Institution Referral Experience. J Urol 2011; 186 (3): 1041–1047. DOI: 10.1016/j.juro.2011.04.099.
- Friedlander DA, Di Carlo HN, Sponseller PD, Gearhart JP. Complications of bladder closure in cloacal exstrophy: Do osteotomy and reoperative closure factor in? J Pediatr Surg 2017; 52 (11): 1836–1841. DOI: 10.1016/j.jpedsurg.2016.12.002.
- Davis R, Sood A, Maruf M, Singh P, Kasprenski MC, DiCarlo HN, et al.. The failed bladder closure in cloacal exstrophy: Management and outcomes. J Pediatr Surg 2019; 54 (11): 2416–2420. DOI: 10.1016/j.jpedsurg.2019.02.012.
- Goldstein SD, Inouye BM, Reddy S, Lue K, Young EE, Abdelwahab M, et al.. Continence in the cloacal exstrophy patient: What does it cost? J Pediatr Surg 2016; 51 (4): 622–625. DOI: 10.1016/j.jpedsurg.2015.12.003.
- Jayman J, Tourchi A, Feng Z, Trock BJ, Maruf M, Benz K, et al.. Predictors of a successful primary bladder closure in cloacal exstrophy: A multivariable analysis. J Pediatr Surg 2019; 54 (3): 491–494. DOI: 10.1016/j.jpedsurg.2018.06.030.
- Inouye BM, Tourchi A, Di Carlo HN, Young EE, Gearhart JP. Modern Management of the Exstrophy-Epispadias Complex. Surg Res Pract 2014; 2014 (587064): 1–9. DOI: 10.1155/2014/587064.
- Shah BB, Di Carlo H, Goldstein SD, Pierorazio PM, Inouye BM, Massanyi EZ, et al.. Initial bladder closure of the cloacal exstrophy complex: Outcome related risk factors and keys to success. J Pediatr Surg 2014; 49 (6): 1036–1040. DOI: 10.1016/j.jpedsurg.2014.01.047.
- Maruf M, Kasprenski M, Jayman J, Goldstein SD, Benz K, Baumgartner T, et al.. Achieving urinary continence in cloacal exstrophy: The surgical cost. J Pediatr Surg 2018; 53 (10): 1937–1941. DOI: 10.1016/j.jpedsurg.2018.02.055.
- Mathews R. Achieving urinary continence in cloacal exstrophy. Semin Pediatr Surg 2011; 20 (2): 126–129. DOI: 10.1053/j.sempedsurg.2010.12.009.
- Surer I, Ferrer FA, Baker LA, Gearhart JP. Continent Urinary Diversion and the Exstrophy-Epispadias Complex. J Urol 2003; 169 (3): 1102–1105. DOI: 10.1097/01.ju.0000044921.19074.d0.
- Woodhouse CRJ, North AC, Gearhart JP. Standing the test of time: long-term outcome of reconstruction of the exstrophy bladder. World J Urol 2006; 24 (3): 244–249. DOI: 10.1007/s00345-006-0053-7.
- Suson K, Novak T, Gupta A, Benson J, Sponseller PD, Gearhart JP. The Neuro-Orthopedic Manifestations of the Omphalocele Exstrophy Imperforate Anus Spinal Defects (OEIS) Complex. J Pediatr Urol 2010; 5 (4): S54. DOI: 10.1016/j.jpurol.2009.02.083.
- Di Carlo HN, Maruf M, Massanyi EZ, Shah B, Tekes A, Gearhart JP. 3-dimensional magnetic resonance imaging guided pelvic floor dissection for bladder exstrophy: a single arm trial. The Journal of Urology 2019; 202 (2): 406–412.
- Tourchi A, Inouye BM, Di Carlo HN, Young E, Ko J, Gearhart JP. New advances in the pathophysiologic and radiologic basis of the exstrophy spectrum. Journal of Pediatric Urology 2014; 10 (2): 212–218.
- Kasprenski M, Michaud J, Yang Z, Maruf M, Benz K, Jayman J, et al.. Urothelial differences in the exstrophy-epispadias complex: potential implications for management. The Journal of Urology 2021; 205 (5): 1460–1465.
Última actualización: 2025-09-21 13:35