
12: 肾脏与尿路先天性畸形
阅读本章大约需要 6 分钟。
引言
肾脏与泌尿道先天性畸形(CAKUT)是一组影响肾脏和泌尿道的结构性异常。据估计,在总体人群中每1万次出生约有4至60例。CAKUT 的诊断通常在常规产前超声检查中作出,或在出生后于具有相关临床体征和症状的新生儿中作出。1 在一项最近报道的纳入409704名婴儿的大型队列中,早产儿的 CAKUT 患病率为2%。2
最常见的畸形是肾盂输尿管连接部梗阻,约影响该病患者的20%。在CAKUT的其他类型中,有多囊性发育不良肾、肾缺如、肾发育不良、肾发育不全、膀胱输尿管返流、巨输尿管、重复肾盂输尿管系统、异位输尿管,以及后尿道瓣膜(图 1)。
尽管在CAKUT的检测和诊断方面已取得显著进展,但该疾病的潜在病因仍未完全明了。需要进一步研究,以加深我们对促成CAKUT发生的胚胎学、遗传学和环境因素的理解,更好地阐明其潜在原因,并改进预防和治疗策略。
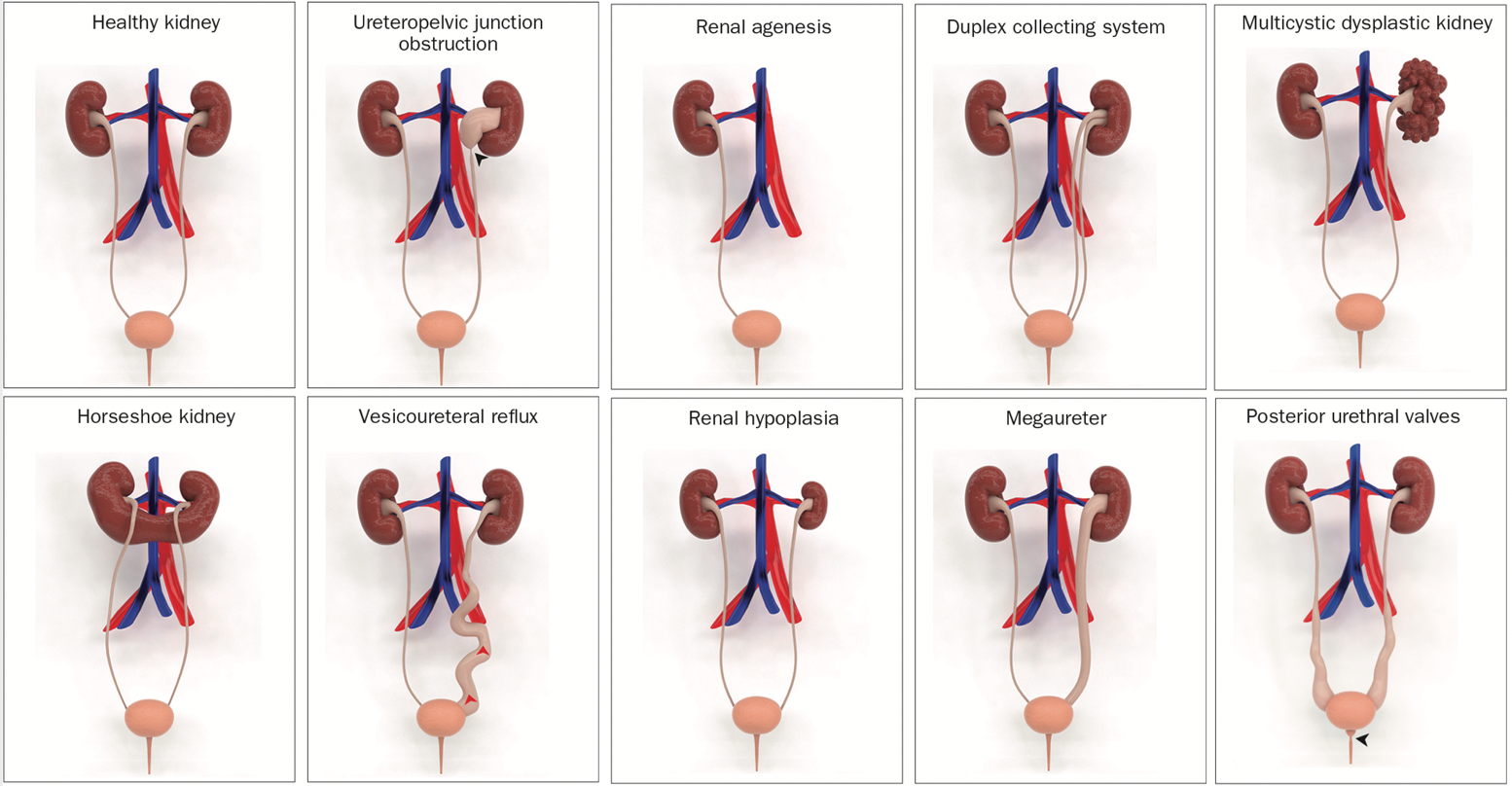
图 1 先天性肾脏和泌尿道畸形(CAKUT)的示意性三维模型。3
胚胎学
胎儿的肾脏和尿路的发育涉及一个复杂的过程,该过程始于妊娠早期的数周,包含多个结构的形成与分化。这个过程始于输尿管芽的形成,输尿管芽源自中间中胚层,产生集合管,并最终形成肾脏。肾脏还由一种称为后肾的结构发育而来,该结构由胚胎中的中胚层形成。
在发育过程中,输尿管芽和后肾必须正确连接并分化,才能形成功能性肾脏和泌尿道。该过程中的紊乱可导致一系列先天性异常。
CAKUT 可按部位(肾脏、输尿管、膀胱或尿道)、按病因(遗传突变、环境暴露、孕期感染等)、按遗传方式以及按严重程度分类(表 1)。在 10% 至 25% 的病例中,CAKUT 归因于遗传性疾病。CAKUT 与遗传性疾病相关,表现出复杂的表型(图 2 和 图 3)。4 具有相同基因组异常的个体可能表现为不同形式的 CAKUT,而相似的表型异常也可能由不同的遗传性疾病引起,并可伴随肾外疾病。2
表 1 CAKUT 疾病类别
| 肾脏 | 集合系统 |
|---|---|
| 数量:肾缺如 | 肾盂输尿管连接部梗阻 |
| 形态:肾发育不全、肾发育不良、多囊性发育不良肾 | 膀胱输尿管返流 |
| 位置:马蹄肾、异位肾、盆腔肾 | 巨输尿管 |
| 重复肾盂输尿管系统 | |
| 后尿道瓣膜 |
多种肾实质畸形可导致肾单位未能正常发育,包括囊性肾发育不良、肾发育不良、肾缺如、肾小管发育不全,以及肾小管发育不全。
多囊肾病
“多囊肾病”一词通常用于描述一种疾病:肾脏内存在多个囊肿,且不伴随相关的发育不良。
常染色体显性多囊肾病 (ADPKD) 和常染色体隐性多囊肾病 (ARPKD) 是最常见的两种多囊肾病类型。除遗传方式(显性或隐性)外,ARPKD 与 ADPKD 在临床表现和预后方面也存在差异。ADPKD 是最常见的 PKD 形式,其由 PKD1 或 PKD2 基因突变所致。
与肾囊性疾病发生相关的基因:5,6
- PKD1: 该基因与ADPKD相关。PKD1基因的突变约占ADPKD病例的85%。
- PKD2: 该基因也与ADPKD相关。PKD2基因的突变约占ADPKD病例的15%。
- HNF1β: 该基因的突变与常染色体显性多囊肾病2型(ADPKD2)有关。
- REN: 该基因与婴儿型多囊肾病有关。
- MUC1: 该基因的突变与一种与MUC1相关的髓质囊性肾病有关。
- INVS: 该基因的突变与与肾单位消耗症相关的髓质囊性肾病有关。
- NPHP1, NPHP3, NPHP4, NPHP5, NPHP6, NPHP7, NPHP8, NPHP9: 这些基因与与肾单位消耗症相关的髓质囊性肾病有关。
- BMP7: 该基因的突变与与肾单位消耗症相关的髓质囊性肾病有关
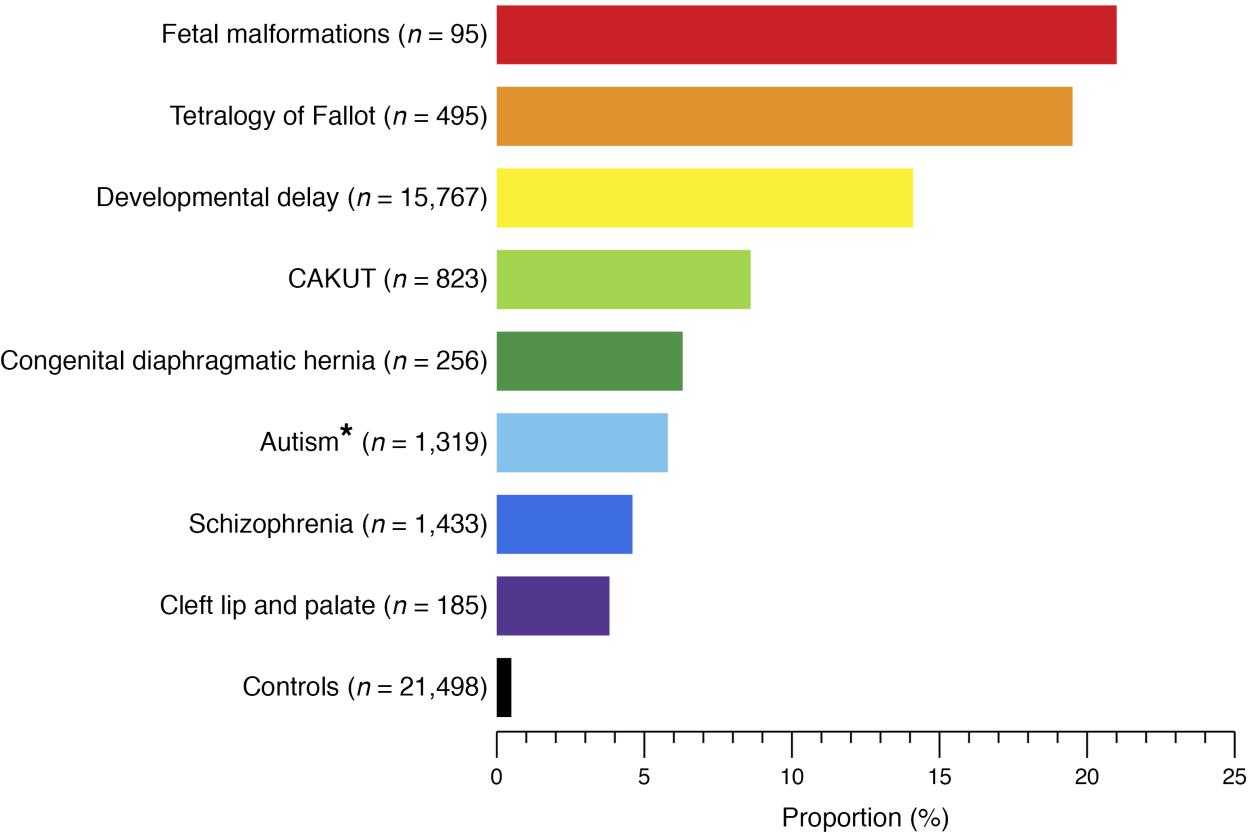
图 2 已知基因组疾病患者在不同人类发育表型和健康对照中的比例。4
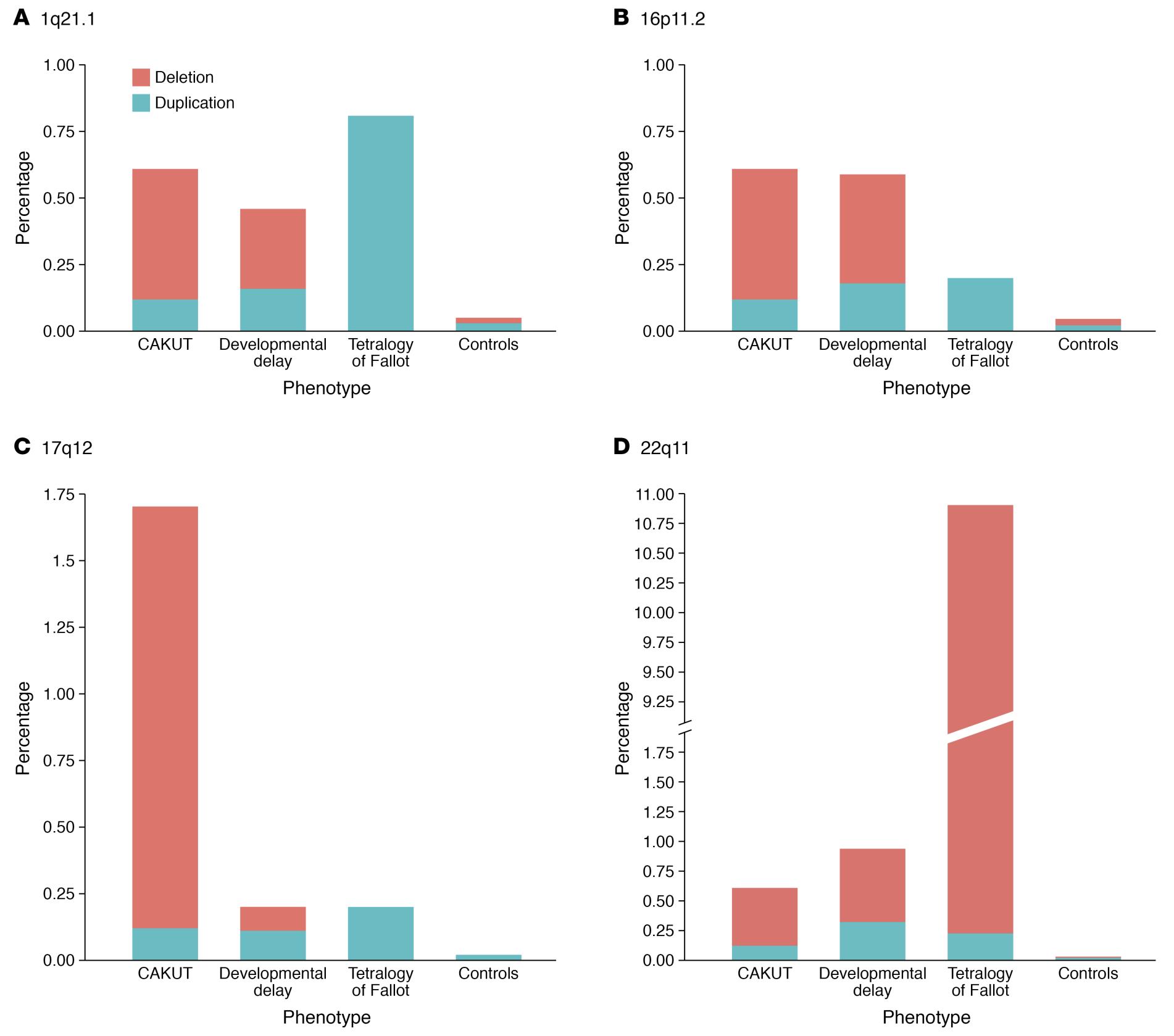
图 3 CAKUT 患者中四个最常涉及的 CNV 位点检出率的差异与相似性。4
多囊性发育不良肾
MCDK 是一种非遗传性的先天性囊性疾病,属于肾发育不良,其特征为多个彼此不相交通的囊肿,由发育不良的实质分隔。MCDK 的发生率约为每 3,600 至 4,300 名活产中有 1 例。7,8 MCDK 的病因尚不明确,但被认为可能与输尿管芽形成异常、暴露于致畸因子或泌尿道梗阻有关。MCDK 常在产前超声检查中被发现。由于其常不引起症状或并发症,出生后通常仅在出现明显的肿块时被识别,或在因其他疾病进行影像学检查时偶然发现。
与MCDK相关的一些基因包括:9,10,11
- HNF1β: 该基因编码一种称为肝细胞核因子1β的蛋白质,在肾脏及其他器官的发育和功能中发挥作用。HNF1β 的突变可导致一系列肾脏异常,包括 MCDK。
- PAX2: 该基因编码一种称为成对盒2的蛋白质,对输尿管的发育很重要,输尿管是将尿液从肾脏输送至膀胱的管道。PAX2 的突变可导致 MCDK 和其他肾脏异常。
- SIX1: 该基因编码一种称为无眼同源盒1的蛋白质,参与包括肾脏在内的多种器官的发育。SIX1 的突变可导致 MCDK 和其他肾脏异常。
- WT1: 该基因编码一种称为 Wilms 肿瘤 1 的蛋白质,对肾脏及其他器官的发育十分重要。WT1 的突变可导致 MCDK 和其他肾脏异常。
MCDK 通常采用保守管理(即观察),因为大多数病例不会出现任何长期并发症。对侧肾异常的患者可能会随着时间出现肾功能受损。因此,需要长期随访。
肾发育缺如
与双侧肾缺如相比,单侧肾缺如在每1,000例妊娠中约发生1例,且预后较好。据估计,每1,000例出生中有0.1%至0.3%受双侧肾缺如影响,其生存率通常较低。在肾缺如家系中已描述了多种遗传模式,肾缺如可作为孤立发现或作为综合征的一部分出现。12
单侧肾缺如可由多个基因的突变所致,包括 ATRX、RET、BMP4、FRAS1、FREM1、GRIP1、GLI3、UPK3A 等。此外,双侧肾缺如可由 RET、FGF20、ITGA8 基因的突变引起。相关基因列于表 2.13,14
肾发育不良
肾发育不全源于胎儿生命第一孕期后肾肾胚芽发育的停滞。然而,其病因仍不明确;宫内血管异常以及遗传性发育异常是两种可能的机制。肾发育不全既可以是孤立发现,也可以是与泌尿道畸形相关的遗传综合征的一部分。ESCAPE研究在一大队列肾发育不全患儿中,对涉及肾发育不良的发育基因(HNF1β、PAX2、EYA1、SIX1 和 SALL1)进行了分析。根据该研究,在具有肾脏病理学改变的患者中,15%检出了 PAX2 突变;而存在大型基因缺失或突变者表现出多变的肾表型。此外,所有囊性肾发育不全患者中有22%存在 HNF1 突变,提示对患有囊性肾发育不良的个体应筛查 HNF1 突变。15 肾脏超声所见为肾体积减小及皮髓质分界消失,临床上提示肾发育不全。随着时间推移,部分肾发育不全患者可进展为终末期肾病。目前尚无针对肾发育不全的特异性治疗。长期随访并管理肾功能十分重要。对于进展为终末期肾病的患者,肾移植是首选方案。
肾盂输尿管连接部
新生儿肾积水最常见的病理原因是输尿管上段的梗阻。总体发生率为1:1,500,新生男婴与女婴之比为2:1.16 UPJ梗阻通常由内在性狭窄引起,较少见的原因是由一条交叉血管在输尿管上段造成的外在压迫。其可能的机制是近端输尿管在胚胎期发生发育中断,改变了环形肌层的发育,即通过改变肌细胞之间及其周围的胶原纤维及其组成。
多种单基因突变被认为与UPJO有关,包括 Id2、PAX2、EYA、AGTR2、BMP4、SOX17、CHD1L 和 DSTYK。17,18 据报道,Id2 和 Adamts1 可产生与人类UPJO相似的表型。缺失 Id2 的小鼠还表现出输尿管高位插入肾盂。19
膀胱输尿管反流
已鉴定出若干与 VUR 发生有关的基因。其中一些基因参与输尿管平滑肌的发育与功能,另一些则参与膀胱的发育与功能。双生子、同胞及子代中 VUR 的高患病率提示该病具有遗传学基础。基于不同人群的遗传学研究结果,已确定 VUR 是一种遗传异质性疾病。一项全基因组连锁与关联研究将原发性非综合征性 VUR 定位到基因组的 10q26 区域。20,21 与 VUR 发生相关的其他基因包括 FOXC1 基因、MYH11 基因和 ACTN4 基因。这些基因在泌尿道的发育与功能中发挥多种作用。22,23 一项采用目前规模最大的 VUR 拷贝数变异分析和全基因组关联研究的全基因组关联研究,鉴定出了五个可能与 VUR 相关、且效应较大的位点,包括 WDPCP、OTX1、BMP5、WDPCP 和 WNT5。WNT5 可能在泌尿生殖系统发育中发挥作用。一项采用目前规模最大的 VUR 拷贝数变异分析和全基因组关联研究的全基因组关联研究,鉴定出了五个可能与 VUR 相关、且效应较大的位点,包括 WDPCP、OTX1、BMP5、WDPCP 和 WNT5。WNT5 可能在泌尿生殖系统发育中发挥作用。24
后尿道瓣膜
后尿道瓣膜是一种危及生命的泌尿道先天异常,通常在新生儿发育过程中被发现;即便接受最佳治疗,仍会导致较高的肾功能不全发生率。PUV 估计在每 7,000–8,000 例活产中发生 1 例。25
有大量报告表明,PUV 是男性新生儿慢性肾病的首要病因,且终末期肾病(ESRD)可发生于5%至64%的病例。26,27 一项系统综述报告称,PUV 患者发生慢性肾病(CKD)和终末期肾脏病(ESKD)的风险最高分别可达32%和20%。28
迄今为止,与后尿道瓣膜相关的基因仅有少数被鉴定出来。梅干腹综合征仅在已识别病例的3%中与_HNF1B_有关联,而大多数确诊者并无遗传学病因。在与21三体相关的CAKUT异常中,包括后尿道瓣膜、肾盂扩张和巨输尿管。29,30
表 2 涉及 CAKUT 发育的基因汇总。31,32,33,22,23,34,35,36
| 疾病 | 基因 |
|---|---|
| 肾发育不全/发育不良 | ACE, AGT, ATRX, CHD7, ESCO2, EYA1, SIX1, FRAS1, FREM2, GATA3, GLI3, GPC3, GRIP1, HNF1β, JAG1, KAL1, LRP4, MKS1–4, NIPBL, PAX2, PBX1, REN, SALL1, SIX5 |
| 肾缺如 | ATRX, BMP4, CBP/EP300, CHD7, ESCO2, FGF20, FRAS1, FREM2, GATA3, GRIP1, GLI3, HNF1β, ITGA8, JAG1, KAL1, KAL2, RET, SALL1, VANGL1 |
| 囊性发育不良 | CBP/EP300, DHCR7, EVC, EVC2, FRAS1, FREM2, GPC-3, HNF1β, JAG1, KAL1, KAL2, MKS1–4, NS1, PEX, PAX2, WT1 |
| 重复集合系统 | NS1 |
| 马蹄肾/异位肾 | GLI3, NIPBL, VANGL1 |
| 肾积水, 肾盂输尿管连接部 | ACE, Adamts-1, AGT, ATRX, BMP4, CHD1L, CHD7, DHCR7, DSTYK, ESCO2, EYA, FRAS1, FREM2, GLI3, HSPG2, JAG1, Id2, NS1, PAX2, PEX, RET, SOX17, VANGL1 |
| 输尿管积水, 巨大输尿管 | CHD7, GLI3 |
| 膀胱输尿管反流 | ATRX, DHCR7, EYA1, FOX1, GATA3, HOXA13, HPSE2, JAG1, KAL1, KAL2, MYH11, NIPBL, PAX2, SALL1, SIX1, SIX5 |
| 后尿道瓣膜 | BNC2, SALL1 |
基因知识的临床重要性及未来影响
随着遗传学评估领域的不断推进,我们很可能会在识别 CAKUT 的遗传学病因以及预测这些疾病的发病可能性方面具备日益增强的能力。这可能促成更早的诊断与治疗,从而改善患者的预后。
需要注意的是,诊断CAKUT并不总是需要进行基因检测。明确诊断通常基于临床表现、病史和影像学检查的综合判断。由于这些结果的解读可能较为复杂并可能影响生育决策,CAKUT的基因检测通常在专科中心于临床遗传学专家的指导下进行。多家实验室(PreventionGenetics、Invitae、Blueprint Genetics等)提供针对多种不同CAKUT疾病的基因检测。其CAKUT检测面板包括对多种与CAKUT相关的基因进行检测,包括 HNF1B、NPHP1 和 UPK1B。针对这些疾病的基因检测有助于确诊、提供疾病严重程度的信息,并有助于遗传咨询和家庭生育计划。值得注意的是,其他基因检测公司和实验室也可能提供CAKUT检测面板。由于CAKUT是一组由不同遗传变异导致的疾病,一些公司的CAKUT基因检测面板可能未涵盖CAKUT的所有遗传致病原因,且不同实验室所覆盖的基因列表可能不同。
对 CAKUT 遗传学组成的全面理解对于制定准确的基因检测策略至关重要。此外,这种理解将有助于指导基因检测中的临床决策。为研究 CAKUT 早期诊断与管理的长期影响,需要开展包括重要临床结局和肾功能在内的纵向研究。
结论与总结
先天性肾脏和泌尿道异常(CAKUT)是影响肾脏和泌尿道的结构性异常。据估计,每10,000次出生中约有4–60例。CAKUT 的诊断通常在产前超声检查期间做出,或在出生后对具有相关临床体征和症状的新生儿作出。最常见的异常是肾盂输尿管连接部梗阻,约影响该病患者的20%。肾脏和泌尿道的发育涉及多个结构的形成与分化,包括输尿管芽和后肾,这些结构必须正确连接,才能形成功能性的肾脏和泌尿道。该过程的紊乱可导致 CAKUT。CAKUT 的疾病类别多样,包括影响肾脏数目、形态和位置的异常,以及影响集合系统的异常。在10–25%的病例中,CAKUT 由遗传性疾病引起。已鉴定出若干与多种肾脏相关疾病发生有关的基因。相关研究仍在进行中,以加深对 CAKUT 病因的认识并制定更好的预防和治疗策略。尽管目前仍存在局限性与困难,分子遗传学诊断在未来或可为改善 CAKUT 患者的临床管理带来希望。
要点
- 肾脏和泌尿道先天性异常(CAKUT)是一组影响肾脏和泌尿道的结构性异常,估计在总体人群中每1万次出生有4至60例。
- 胎儿肾脏和泌尿道的发育是一个复杂过程,始于妊娠早期数周,涉及多种结构的形成与分化。该过程的中断可导致一系列先天性异常。
- 在一项近期的大型队列(409,704名婴儿)报告中,早产儿CAKUT的患病率为2%。
- 最常见的异常是肾盂输尿管连接部梗阻,约影响20%的该病患者。
- 在10-25%的病例中,CAKUT可归因于遗传性疾病。
- 遗传学评估正在推进,提高了识别CAKUT遗传原因并预测发生这些疾病可能性的能力。
- 诊断CAKUT并不总是需要基因检测,通常基于临床表现、病史和影像学检查的综合判断。
- 全面理解CAKUT的遗传组成对于制定准确的基因检测策略并指导基因检测中的临床决策至关重要。
- 需要开展纵向研究,以探讨CAKUT早期诊断与管理的长期影响。
参考文献
- Murugapoopathy V, Gupta IR. A Primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin J Am Soc Nephrol 2020; 15 (5): 723–731. DOI: 10.2215/cjn.12581019.
- Hays T, Thompson MV, Bateman DA, Sahni R, Tolia VN, Clark RH, et al.. The Prevalence and Clinical Significance of Congenital Anomalies of the Kidney and Urinary Tract in Preterm Infants. JAMA Netw Open 2022; 5 (9): e2231626. DOI: 10.1001/jamanetworkopen.2022.31626.
- Nicolaou N, Renkema KY, Bongers EMHF, Giles RH, Knoers NVAM. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol 2015; 11 (12): 720–731. DOI: 10.1038/nrneph.2015.140.
- Srivastava S, Molinari E, Raman S, Sayer JA. Many Genes–One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front Pediatr 2018; 5. DOI: 10.3389/fped.2017.00287.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. 8 (1): 25–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 2018; 128 (1): 4–15. DOI: 10.1172/jci95300.
- Gordon AC, Thomas DF, Arthur RJ, Irving HC. Multicystic dysplastic kidney: Is nephrectomy still appropriate? J Pediatr Surg 1988; 24 (9): 951. DOI: 10.1016/s0022-3468(89)80673-6.
- Schreuder MF, Westland R, Wijk JAE van. Unilateral multicystic dysplastic kidney: a meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol Dial Transplant 2009; 24 (6): 1810–1818. DOI: 10.1093/ndt/gfn777.
- Kopač M, Kordič R. Associated Anomalies and Complications of Multicystic Dysplastic Kidney. Pediatr Rep 2022; 14 (3): 375–379. DOI: 10.3390/pediatric14030044.
- Fletcher J, Hu M, Berman Y, Collins F, Grigg J, McIver M, et al.. Multicystic Dysplastic Kidney and Variable Phenotype in a Family with a Novel Deletion Mutation ofPAX2. J Am Soc Nephrol 2005; 16 (9): 2754–2761. DOI: 10.1681/asn.2005030239.
- Chang Y-M, Chen C-C, Lee N-C, Sung J-M, Chou Y-Y, Chiou Y-Y. PAX2 Mutation-Related Renal Hypodysplasia: Review of the Literature and Three Case Reports. Front Pediatr 2022; 9 (765929). DOI: 10.3389/fped.2021.765929.
- Bienstock JL, Birsner ML, Coleman F, Hueppchen NA. Successful In Utero Intervention for Bilateral Renal Agenesis. Obstet Gynecol 2014; 124 (2): 413–415. DOI: 10.1097/aog.0000000000000339.
- Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, et al.. Faculty Opinions recommendation of Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2012; 1 (2): 96–200. DOI: 10.3410/f.13934957.15391059.
- Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal Aplasia in Humans Is Associated with RET Mutations. Am J Hum Genet 2008; 82 (2): 344–351. DOI: 10.1016/j.ajhg.2007.10.008.
- Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al.. Faculty Opinions recommendation of Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2006; 7 (10): 864–870. DOI: 10.3410/f.1047767.508721.
- LEBOWITZ ROBERTL, GRISCOM NTHORNE. Neonatal Hydronephrosis. Radiol Clin North Am 1977; 15 (1): 49–59. DOI: 10.1016/s0033-8389(22)02539-8.
- AC SJ, DM M, oes S, AC S. Congenital Anomalies of the Kidney and Urinary Tract: An Overview. Congenital Anomalies of the Kidney and Urinary Tract 2014; 02 (4): 1–13. DOI: 10.1007/978-3-319-29219-9_1.
- Avanoglu A, Tiryaki S. Embryology and Morphological (Mal)Development of UPJ. Front Pediatr 2020; 8: 137. DOI: 10.3389/fped.2020.00137.
- Aoki Y, Mori S, Kitajima K, Yokoyama O, Kanamaru H, Okada K, et al.. Id2 haploinsufficiency in mice leads to congenital hydronephrosis resembling that in humans. Genes Cells 2004; 9 (12): 1287–1296. DOI: 10.1111/j.1365-2443.2004.00805.x.
- Williams G, Fletcher JT, Alexander SI, Craig JC. Vesicoureteral reflux. Journal of the American Society of Nephrology 2008; 19 (5): 847–862. DOI: 10.1681/asn.2007020245.
- Darlow JM, Darlay R, Dobson MG, Stewart A, Charoen P, Southgate J, et al.. Genome-wide linkage and association study implicates the 10q26 region as a major genetic contributor to primary nonsyndromic vesicoureteric reflux. Sci Rep 2017; 7 (1): 59. DOI: 10.1038/s41598-017-15062-9.
- Woolf AS, Lopes FM, Ranjzad P, Roberts NA. Congenital Disorders of the Human Urinary Tract: Recent Insights From Genetic and Molecular Studies. Front Pediatr 2019; 7 (136). DOI: 10.3389/fped.2019.00136.
- Wu C-HW, Mann N, Nakayama M, Connaughton DM, Dai R, Kolvenbach CM, et al.. Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT). Genet Med 2020; 22 (10): 1673–1681. DOI: 10.1038/s41436-020-0844-z.
- Verbitsky M, Krithivasan P, Batourina E, Khan A, Graham SE, Marasà M, et al.. Review of: "Genome-wide association study followed by trans-ancestry meta-analysis identify 17 new risk loci for schizophrenia". J Am Soc Nephrol 2021; 32 (4): 805–820. DOI: 10.32388/jqb9pr.
- Thakkar D, Deshpande AV, Kennedy SE. Epidemiology and demography of recently diagnosed cases of posterior urethral valves. Pediatr Res 2014; 76 (6): 560–563. DOI: 10.1038/pr.2014.134.
- Warshaw BL, Edelbrock HH, Ettenger RB, Malekzadeh MH, Pennisi AJ, Uittenbogaart CH, et al.. Renal transplantation in children with obstructive uropathy. J Pediatr Surg 1980; 15 (6): 986. DOI: 10.1016/s0022-3468(80)80355-1.
- Roth KS, Carter WH, Chan JCM. Obstructive Nephropathy in Children: Long-Term Progression After Relief of Posterior Urethral Valve. Pediatrics 2001; 107 (5): 1004–1010. DOI: 10.1542/peds.107.5.1004.
- Hennus PML, Kort LMO de, Bosch JLH, Jong TPVM de, Heijden GJMG van der. A Systematic Review on the Accuracy of Diagnostic Procedures for Infravesical Obstruction in Boys. PLoS One 2014; 9 (2): e85474. DOI: 10.1371/journal.pone.0085474.
- Rodriguez MM. Congenital anomalies of the kidney and urinary tract (CAKUT). Lijec Vjesn 2014; 144 (Supp 1). DOI: 10.26800/lv-144-supl1-26.
- Postolache L, Parsa A, Simoni P, Boitsios G, Ismaili K, Schurmans T, et al.. Widespread kidney anomalies in children with Down syndrome. Pediatr Nephrol 2022; 37 (10): 2361–2368. DOI: 10.1007/s00467-022-05455-y.
- Uy N, Reidy K. Developmental Genetics and Congenital Anomalies of the Kidney and Urinary Tract. J Pediatr Genet 2016; 05 (01): 051–060. DOI: 10.1055/s-0035-1558423.
- Kolvenbach CM, Dworschak GC, Frese S, Japp AS, Schuster P, Wenzlitschke N, et al.. Expanding congenital abnormalities of the kidney and urinary tract (CAKUT) genetics: basonuclin 2 (BNC2) and lower urinary tract obstruction. Ann Transl Med 2019; 7 (S6): S226–s226. DOI: 10.21037/atm.2019.08.73.
- Heidet L, Morinière V, Henry C, De Tomasi L, Reilly ML, Humbert C, et al.. Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 2017; 28 (10): 2901–2914. DOI: 10.1681/asn.2017010043.
- Talati AN, Webster CM, Vora NL. Prenatal genetic considerations of congenital anomalies of the kidney and urinary tract (CAKUT). Prenat Diagn 2019; 39 (9): 679–692. DOI: 10.1002/pd.5536.
- Puri P, Gosemann J-H, Darlow J, Barton DE. Genetics of vesicoureteral reflux. Nat Rev Urol 2011; 8 (10): 539–552. DOI: 10.1038/nrurol.2011.113.
- Chen F. Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol 2009; 24 (9): 1621–1632. DOI: 10.1007/s00467-008-1072-y.
最近更新时间: 2025-09-22 08:00