
40: DSD—Compreensão atual, avaliação diagnóstica e tratamento
Este capítulo levará aproximadamente 23 minutos para ler.
Introdução e Terminologia
Distúrbios/diferenças do desenvolvimento sexual (DSD, também referidos como intersexo) são condições congênitas nas quais o sexo cromossômico, gonadal ou fenotípico é diferente do que se considera tipicamente masculino ou feminino. Uma nova e ampla classificação de DSD foi introduzida em 2006 por meio do “Consensus Statement on Management of Intersex Disorders.”1 Essa classificação mais recente incorpora uma ampla variedade de condições, incluindo hiperplasia adrenal congênita (HAC), DSD ovotesticular, síndrome da insensibilidade aos andrógenos (SIA). A definição de consenso de 2006 é mais abrangente do que o que anteriormente era referido como condições intersexo, incluindo também anomalias anatômicas como extrofia cloacal e vesical e agenesia vaginal, e anomalias cromossômicas que não causam genitália atípica (por exemplo, Síndrome de Klinefelter [47, XXY]). Tabela 1 fornece uma visão geral da nomenclatura anterior e das atualizações de terminologia recomendadas propostas na declaração de consenso de 2006. Dada a ampla gama de condições sob o ‘guarda-chuva’ DSD, uma abordagem individualizada e multidisciplinar de cuidado é primordial.
Tabela 1 Nomenclatura de DSD revisada. Fonte: Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
| Anterior | Proposto |
|---|---|
| Intersexo | DSD |
| Pseudo-hermafrodita masculino | 46,XY DSD |
| Pseudo-hermafrodita feminino | 46,XX DSD |
| Hermafrodita verdadeiro | Ovotesticular DSD |
| Disgenesia gonadal mista | Disgenesia gonadal mista (inalterada) |
| Homem XX ou reversão sexual XX | 46,XX testicular DSD |
| Reversão sexual XY | 46,XY disgenesia gonadal completa |
Houve uma adoção quase universal da nova terminologia de DSD pela comunidade médica desde que foi introduzida. Entre indivíduos afetados por condições de DSD, existe variabilidade nas preferências de terminologia, e falta consenso sobre exatamente quais condições devem ser consideradas “um DSD”.2,3,4 Em particular, alguns membros da comunidade de CAH se identificam como tendo um distúrbio endócrino, não uma condição de DSD/intersexo.5 Também não está claro quais indivíduos com hipospádia proximal devem ser considerados como tendo “um DSD”.6 Clínicos que tratam condições de DSD devem estar cientes da evolução e das controvérsias da nomenclatura e usar os termos que seus pacientes preferem durante atendimentos médicos individuais. Para os fins deste capítulo, será utilizada a terminologia atual, aceita pela comunidade médica.
Embriologia
Desenvolvimento típico da genitália interna/externa
Todos os fetos em desenvolvimento começam da mesma forma. As estruturas anatômicas para o desenvolvimento masculino ou feminino e suas variações estão presentes nas primeiras semanas de gestação (Tabela 2), incluindo a crista gonadal, os ductos de Wolff (mesonéfricos) e de Müller (paramesonéfricos), a cloaca e o subsequente seio urogenital, o tubérculo genital e as saliências lábio-escrotais. Figura 1 mostra genes responsáveis pelo desenvolvimento da gônada indiferenciada em testículo ou ovário.
Tabela 2 Precursores embriológicos e estruturas típicas masculinas/femininas.
| Estrutura embriológica | Estrutura típica feminina | Estrutura típica masculina |
|---|---|---|
| Crista gonadal | Ovário | Testículo |
| Ducto de Wolff (mesonéfrico) | Paroóforo, epoóforo, cisto do ducto de Gartner | Ducto deferente, epidídimo, vesículas seminais |
| Ducto de Müller (paramesonéfrico) | Trompas de Falópio, útero, vagina proximal | Apêndice testicular, utrículo prostático |
| Cloaca e subsequente seio urogenital | Bexiga, vagina distal, uretra | Bexiga, próstata, uretra |
| Tubérculo genital | Clitóris | Pênis |
| Intumescências lábio-escrotais | Complexo labial | Escroto |
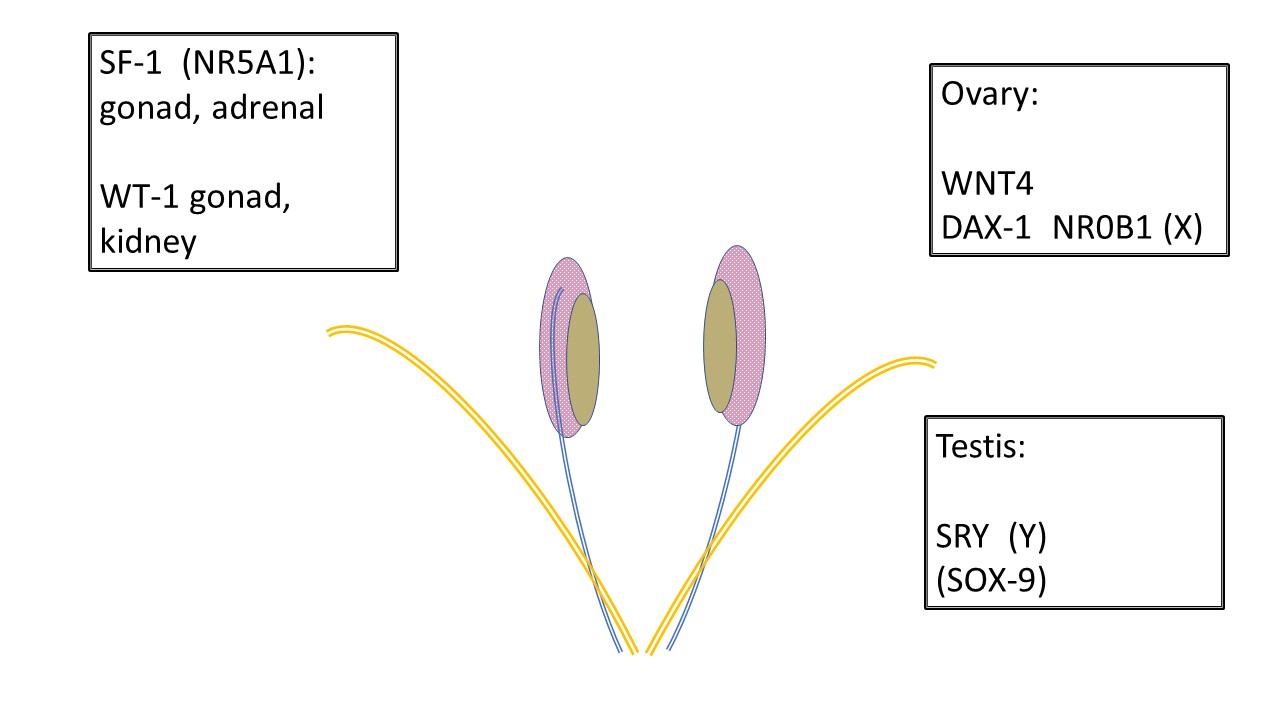
Figura 1 Desenvolvimento de gônadas indiferenciadas. Todas as gônadas começam da mesma forma. Células germinativas primordiais migram para a crista gonadal antes de 6 semanas e a infraestrutura para sustentar o desenvolvimento gonadal é ainda influenciada por vários genes. SF-1 e WT-1 têm influência sobre o desenvolvimento gonadal e a subsequente comunicação endócrina para os ductos de Wolff (mesonéfrico-azul) e de Müller (paramesonéfrico-laranja). Observe a relação entre o mesonefro (rosa), o ducto mesonéfrico e a gônada indiferenciada (bege). WNT4 é o gene determinante do ovário. DAX-1 é considerado o “gene anti-testicular.” A duplicação de DAX-1 resulta em inversão sexual XY. SRY no cromossomo Y é o gene determinante do testículo. SOX-9 favorece o desenvolvimento das células de Sertoli, mas também apresenta homologia para participar do desenvolvimento testicular na ausência de SRY.
O desenvolvimento masculino típico (Figura 2) é iniciado muito precocemente na gestação pelo cromossoma Y, especificamente o SRY, determinando o desenvolvimento testicular. WT-1, SF-1 estão envolvidos no desenvolvimento do testículo e do ovário, e SOX-9 está envolvido na diferenciação das células de Sertoli. As células de Leydig do testículo iniciam posteriormente a produção de testosterona por volta das 8 semanas de gestação e as células de Sertoli produzem a hormona anti-Mülleriana (AMH), inibindo a maturação dos ductos de Müller em útero, trompas de Falópio e porção superior da vagina. A testosterona determina o crescimento inicial do tubérculo genital e o desenvolvimento do ducto de Wolff em ducto deferente, epidídimo e vesícula seminal. O desenvolvimento, ou a sua ausência, nos sistemas de Wolff e de Müller ocorre como uma ação parácrina e não sistémica, o que explica o desenvolvimento assimétrico das estruturas ductais quando a composição da gónada é assimétrica. A conversão da testosterona no mais potente diidrotestosterona (DHT) pela 5α-redutase-2 ocorre em vários tecidos e é responsável pela maturação adicional do pénis, pela tubularização do seio urogenital e da placa uretral, e pelo fecho do escroto até às 14 semanas de gestação. O crescimento subsequente da estrutura fálica ocorre devido ao declínio dos androgénios fetais e ao crescimento somático.
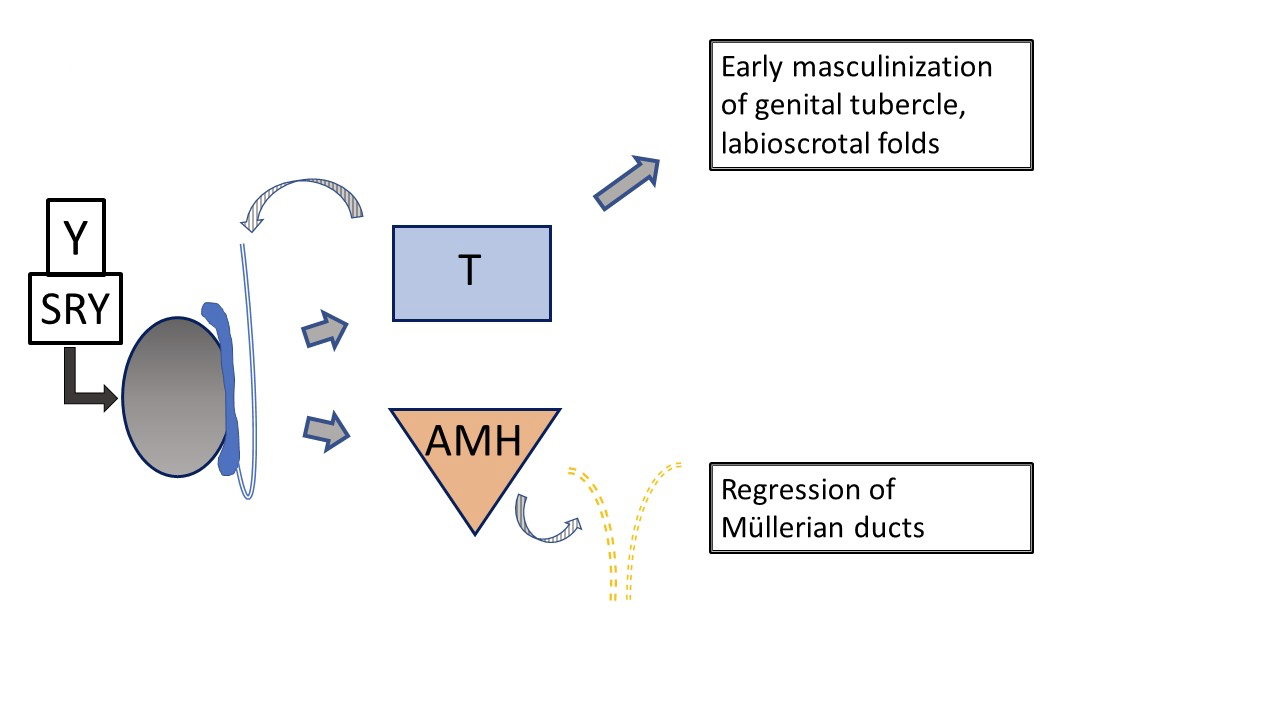
Figura 2 Desenvolvimento inicial típico do trato genital externo e interno masculino, influenciado pelo testículo fetal. A produção de testosterona e AMH pelo testículo fetal resulta em masculinização precoce do tubérculo genital e regressão dos ductos de Müller. O desenvolvimento subsequente da genitália externa e da uretra é influenciado pela conversão da testosterona em dihidrotestosterona. (não representado aqui, ver Figura 4).
Desenvolvimento feminino típico (Figure 3) é frequentemente considerado como ocorrendo por defeito, uma vez que não é regulado por hormonas produzidas pelos testículos. A determinação ovárica não tem papel de destaque no desenvolvimento embriológico da genitália. Níveis baixos de androgénios e AMH permitem o desenvolvimento feminino típico do tubérculo genital como clitóris, das pregas lábio-escrotais como complexo labial e do seio urogenital distal como uretra e vagina. Os ductos de Müller pareados migram e fundem-se para formar o útero. Em conjunto, estimulam o bulbo sinovaginal no seio urogenital a iniciar o desenvolvimento da porção inferior da vagina.
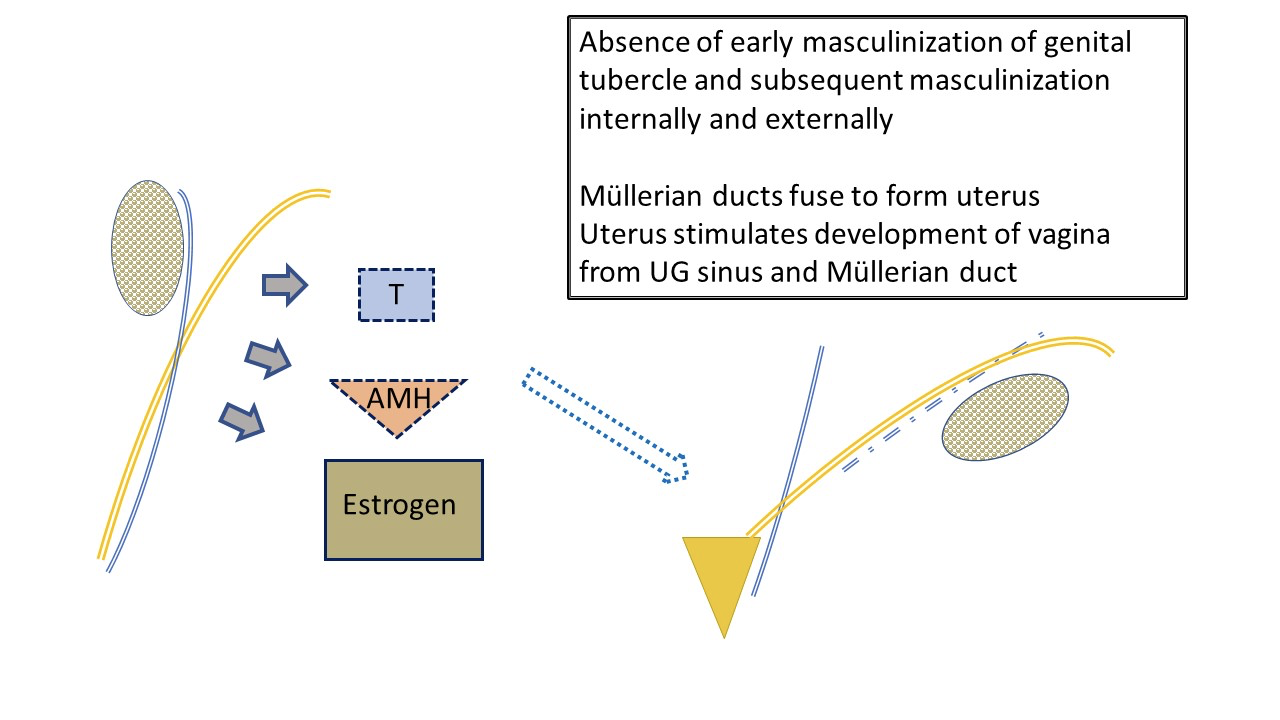
Figura 3 Desenvolvimento inicial típico do trato genital feminino externo e interno, influenciado pelo ovário fetal. Baixos níveis de AMH e de andrógenos pelo ovário resultam no desenvolvimento da anatomia feminina típica: útero e trompas de Falópio, porção superior da vagina. A produção fetal de estrogénios tem menos impacto no desenvolvimento da linha feminina do que um ambiente com níveis mínimos de andrógenos e AMH. Os lábios serão afetados pelos estrogénios maternos. Note também a relação final de “água debaixo da ponte” entre o uréter (ramificado do ducto mesonéfrico) e a trompa de Falópio. Note os remanescentes do ducto mesonéfrico associados à trompa de Falópio.
Diferentes vias que podem causar DSD
Diferenças no desenvolvimento podem ocorrer por meio de atipias:
- Determinação genética das gônadas (p. ex., disgenesia gonadal mista, DSD ovotesticular)
- Produção hormonal (p. ex., deficiência de 5α-redutase, deficiência de 3-β-hidroxiesteroide desidrogenase, CAH por deficiência de 21-hidroxilase, ou mutações do receptor do Hormônio Luteinizante [LH])
- Ação hormonal (p. ex., AIS completa ou parcial)
- Variações no desenvolvimento do tecido precursor/separação da cloaca (p. ex., síndrome de Mayer-Rokitansky-Küster-Hauser [MRKH], extrofia cloacal, anomalias cloacais)
Mesmo com um cariótipo 46,XX ou 46,XY, não há certeza de que o desenvolvimento seguirá a linha feminina ou masculina típica. Somente em 46,XY, ocorrem inúmeras diferenças devido a mutações no cromossomo Y que alteram a determinação ou a função testicular, mutações somáticas que levam à disgênese do testículo em desenvolvimento, variações na biossíntese ou na conversão da testosterona e mutações ligadas ao X que afetam a função do receptor de andrógenos. Fenótipo externo, desenvolvimento interno, potencial para puberdade espontânea ou fertilidade e identidade ao longo do espectro de gênero podem todos ser afetados. Ao considerar as características das várias condições de DSD descritas abaixo, é importante observar que a produção e a ação hormonais ocorrem em um espectro. Assim, deficiências na função do receptor ou na produção de hormônio e de enzimas de conversão não são necessariamente déficits completos. O fenótipo pode variar amplamente, mesmo entre indivíduos com o mesmo diagnóstico.
Fisiopatologia
Variações no desenvolvimento da genitália interna e externa são orientadas por mutações dos cromossomos sexuais e mutações somáticas, pela função gonadal resultante e pela resposta tecidual. Quando a função do receptor ou a presença de uma enzima pertinente é atípica devido a uma mutação genética conhecida ou ainda não identificada, o efeito sobre os órgãos-alvo pode ser completo (sem função evidente) ou parcial. O fenótipo varia de acordo. Como exemplo, vários fenótipos de insensibilidade aos andrógenos (CAIS completa, PAIS parcial e MAIS leve: espectro do fenótipo genital de feminino típico—ambíguo—masculino típico) ocorrem por meio de numerosas mutações distintas do gene do Receptor de Andrógeno no Cromossomo X (condição recessiva ligada ao X). Tabela 3 mostra um sistema de classificação de DSD, dividido por cariótipo.
Tabela 3 Um Exemplo de uma Classificação de DSD
| DSD dos cromossomos sexuais | DSD 46,XY | DSD 46,XX |
|---|---|---|
| 45,X (síndrome de Turner e variantes) | Distúrbios do desenvolvimento gonadal (testicular): (1) disgênese gonadal completa (síndrome de Swyer); (2) disgênese gonadal parcial; (3) regressão gonadal; e (4) DSD ovotesticular |
Distúrbios do desenvolvimento gonadal (ovariano): (1) DSD ovotesticular; (2) DSD testicular (por exemplo, SRY+, duplicação de SOX9); e (3) disgênese gonadal |
| 47,XXY (síndrome de Klinefelter e variantes) | Distúrbios na síntese ou ação dos andrógenos: (1) defeito na biossíntese de andrógenos (por exemplo, deficiência de 17-hidroxiesteroide desidrogenase, deficiência de 5RD2, mutações em StAR); (2) defeito na ação dos andrógenos (por exemplo, CAIS, PAIS); (3) defeitos do receptor do hormônio luteinizante (por exemplo, hipoplasia de células de Leydig, aplasia); e (4)distúrbios do hormônio anti-Mülleriano e do receptor do hormônio anti-Mülleriano (síndrome de persistência dos ductos de Müller) |
Excesso de andrógenos: (1) fetal (por exemplo, deficiência de 21-hidroxilase, deficiência de 11-hidroxilase); (2) fetoplacentário (deficiência de aromatase, POR [Oxidorredutase P450]); e (3) materno (luteoma, exógeno, etc) |
| 45,X/46,XY (MGD, DSD ovotesticular) | Outros (por exemplo, extrofia cloacal, atresia vaginal, MURCS [anomalias müllerianas, renais, dos somitos cervicotorácicos], outras síndromes) | |
| 46,XX/46,XY (quimérico, DSD ovotesticular) |
46,XX DDS
A forma mais comum de DSD 46,XX é a CAH, na qual a função enzimática adrenal está comprometida. Figura 4 mostra a cascata típica de esteroides adrenais, que se inicia com o colesterol. Os produtos finais são aldosterona, cortisol e esteroides sexuais. O ramo da cascata referente aos esteroides sexuais termina com a androstenediona, que é convertida em uma pequena quantidade de testosterona. A produção de testosterona nas células de Leydig do testículo também se inicia com o colesterol. Na CAH, a função enzimática reduzida causa diminuição dos mineralocorticoides (aldosterona) e dos glicocorticoides (cortisol), e um desvio para a produção de esteroides sexuais. O resultado é a masculinização de um feto com cariótipo 46,XX. Embora seja mais fácil lembrar da ausência completa de função enzimática e dos produtos esteroides adrenais subsequentes, a função pode estar apenas parcialmente comprometida e a retroalimentação no eixo hipotálamo-hipofisário ser afetada de modo correspondente.
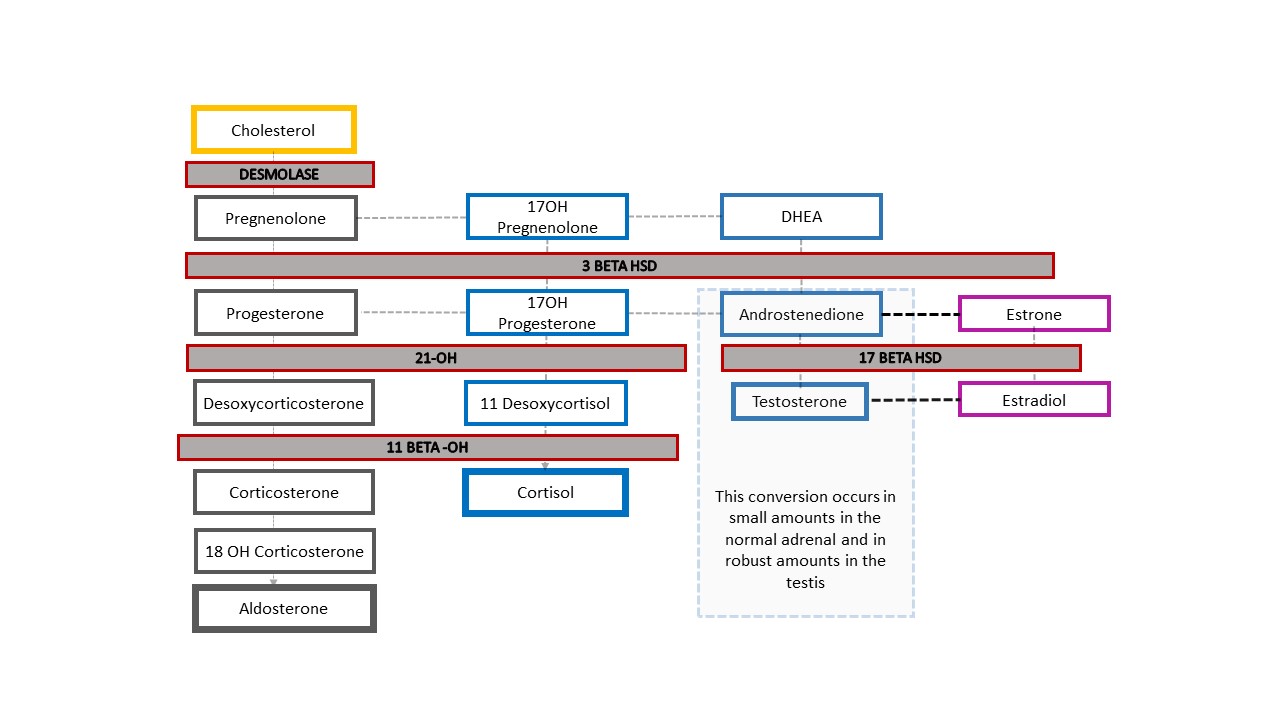
Figura 4 Cascata de esteroides e espectro dos diagnósticos de HAC. Qualquer grau de defeito na produção de uma enzima resulta em diminuição dos produtos a jusante e desvio da cascata em direção aos esteroides sexuais. O eixo hipotálamo-hipófise-adrenal é estimulado para compensar. O aumento de ACTH resulta em hiperplasia do córtex adrenal e aumento adicional da produção de precursores.
A anatomia típica de um paciente com HAC 46,XX inclui graus variáveis de aumento do clítoris e formação de seio urogenital. As gónadas são ovários e não são palpáveis. Existem várias mutações que levam a diferentes perfis de esteroides adrenais característicos descritos nos manuais e a fenótipos físicos e fisiológicos. A forma mais comum é a deficiência de 21-hidroxilase. Recém-nascidos com HAC 46,XX podem apresentar perda salina com risco de vida por volta de uma semana de vida; portanto, este diagnóstico deve ser considerado para todos os recém-nascidos com genitália ambígua. Uma 17-OH-progesterona elevada é diagnóstica. A identidade de género feminina e o sexo de criação feminino são mais frequentes, embora não universais.
46,XY DSD
Dentro da categoria de DSD 46,XY, há uma gama de distúrbios do desenvolvimento gonadal e da síntese e ação de andrógenos, conforme detalhado na Tabela 3. Juntamente com o espectro de condições de AIS detalhadas acima, a disgenesia gonadal completa ou parcial pode resultar em variações anatômicas semelhantes: um lactente com cromossomos 46,XY e fenótipo feminino típico ou ambíguo (por exemplo, hipospádia proximal com transposição penoescrotal, e uma ou ambas as gônadas não descidas). Indivíduos com AIS completa e disgenesia gonadal completa são tipicamente criados como do sexo feminino e se identificam como do sexo feminino; aqueles com formas parciais de qualquer uma das condições apresentam maior variabilidade no sexo de criação e na identidade de gênero.
Outra DDS 46,XY, a deficiência de 5α-redutase, causa conversão reduzida de testosterona em di-hidrotestosterona (DHT), resultando em fenótipo genital variável. Figura 5 e Figura 6 comparam a via típica de desenvolvimento testicular com a via que ocorre em pacientes com deficiência de 5α-redutase.
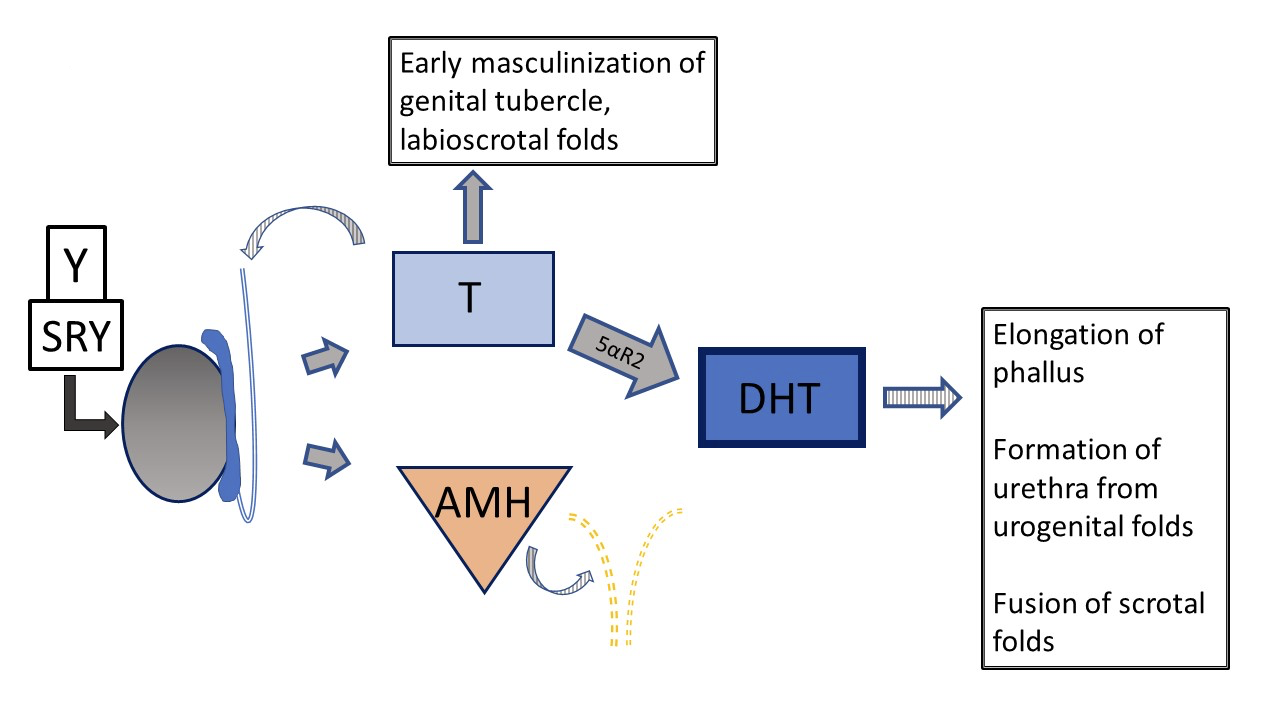
Figura 5 Desenvolvimento genital pré-natal típico 46XY O desenvolvimento pode desviar-se do “típico” em várias etapas diferentes. Setas listradas significam função do tipo parácrino. Linhas tracejadas indicam regressão dos ductos Müllerianos no início do desenvolvimento.
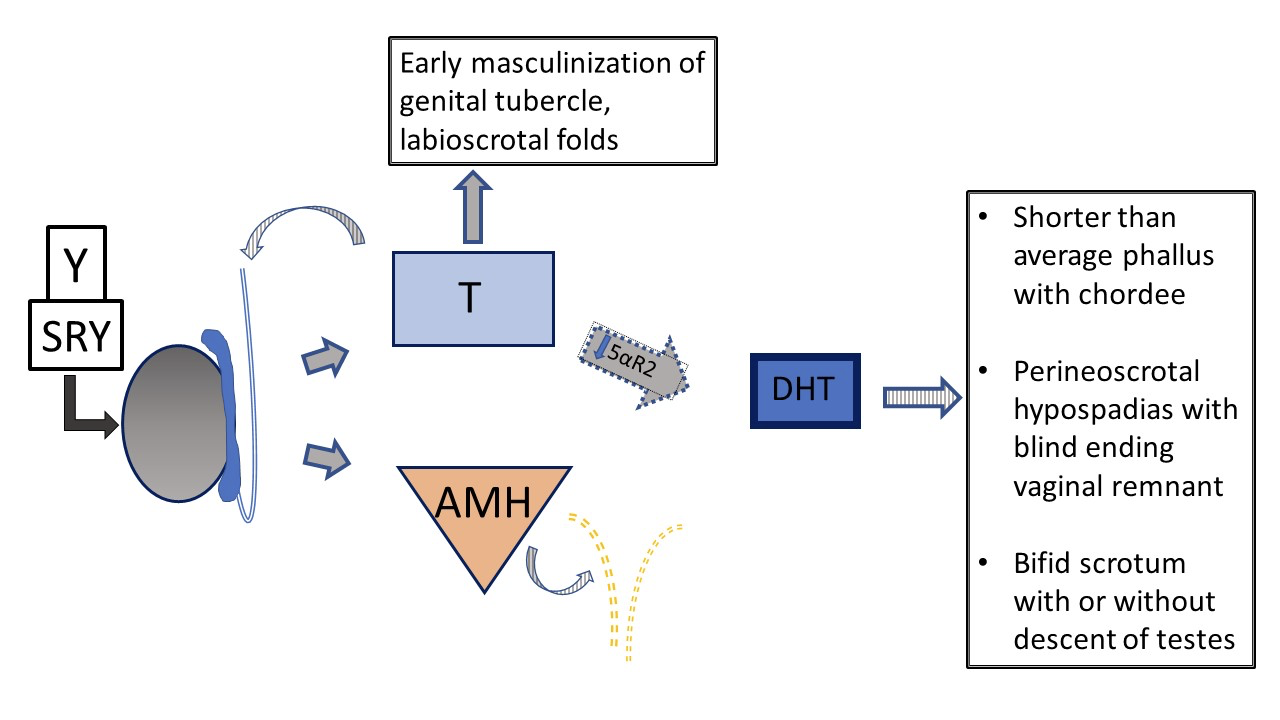
Figura 6 Desenvolvimento genital pré-natal 46XY em pacientes com vs deficiência de 5-α-redutase. Os testículos são determinados e apresentam produção normal de testosterona e AMH. Não há útero. Devido à função comprometida da 5-α-redutase, a relação T /DHT está marcadamente aumentada. No contexto de DHT reduzida, há variabilidade no fenótipo, do feminino típico ao masculino típico, até anatomia genital ambígua. Com deficiência grave, o desenvolvimento do falo e do seio urogenital é interrompido em estágio inicial de masculinização, com um único orifício urogenital na região perineal e sem fusão das pregas lábio-escrotais.
Figura 7 mostra a cascata adrenal resultante no exemplo raro de um paciente com HAC 46,XY que se apresenta clinicamente com genitália ambígua: deficiência de 3-β-hidroxiesteroide desidrogenase [HSD]. O bloqueio enzimático ocorre antes da conversão de desidroepiandrosterona (DHEA) em androstenediona na cascata esteroidal adrenal, de modo que lactentes apresentam mineralocorticoides e glicocorticoides reduzidos, juntamente com uma diferença no desenvolvimento genital. Como bebês com HAC 46,XX e deficiência de 21-hidroxilase ou 11β-hidroxilase, lactentes com deficiência de 3-β-HSD podem apresentar perda de sal com risco de vida. O diagnóstico é feito por meio de 17-OH pregnenolona e DHEA elevados.
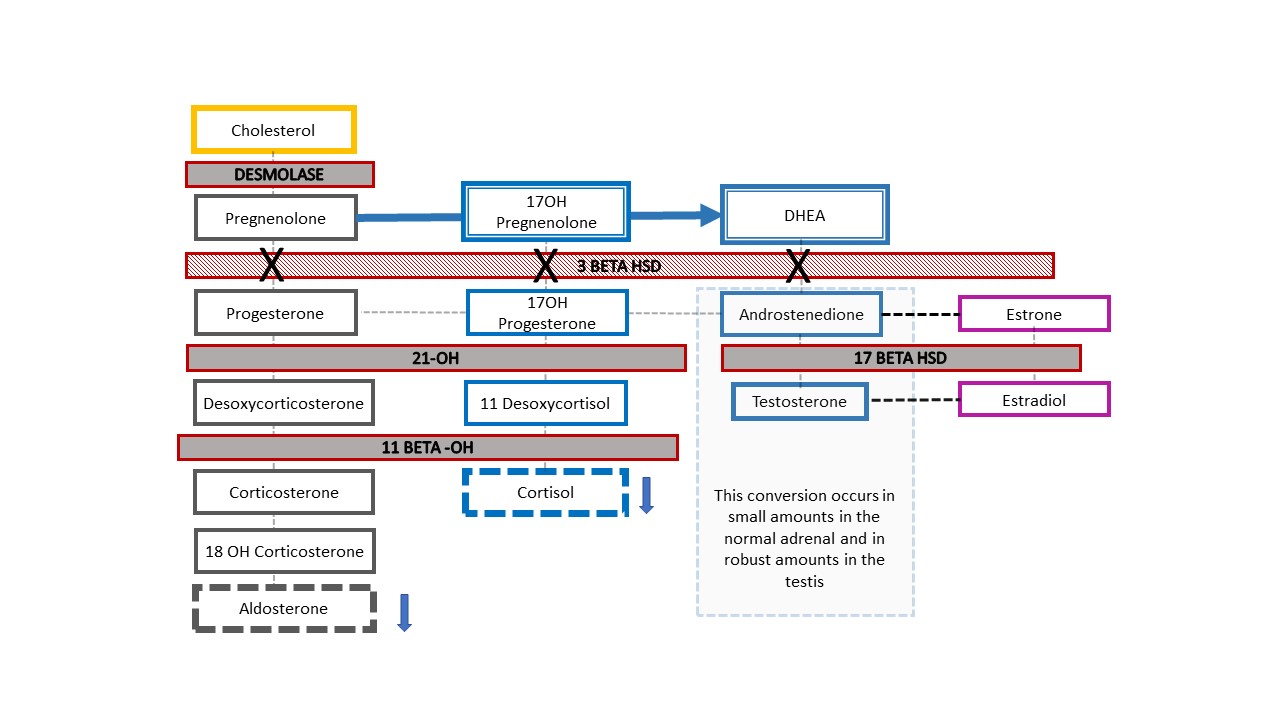
Figura 7 Via esteroidal na deficiência da 3-β-hidroxiesteroide desidrogenase. Este defeito relativamente proximal na cascata do colesterol adrenal resulta em deficiência grave de corticosteroides e mineralocorticoides. É singular, pois bebês 46,XY com HAC apresentam aspecto genital hipomasculinizado, enquanto bebês 46,XX têm aspecto genital feminino típico (ou quase típico). Quando um recém-nascido com cromossomo Y detectado no pré-natal nasce com aspecto ambíguo da genitália, este é o diagnóstico de HAC a ser excluído. Níveis elevados de 17-OH-pregnenolona e DHEA ajudam a estabelecer o diagnóstico.
DSD dos cromossomos sexuais
Dos diagnósticos de DSD dos cromossomos sexuais listados na Tabela 3, as condições com cariótipo mosaico 45,X/46,XY (ou semelhante) merecem menção especial. Pacientes com esse cariótipo podem apresentar genitália externa de aspecto feminino típico (Síndrome de Turner com material do cromossomo Y [TS+Y]), genitália ambígua (disgenesia gonadal mista [MGD]) ou genitália externa de aspecto masculino típico. Pacientes com TS+Y têm risco aumentado de tumores gonadais, diferindo dos pacientes com síndrome de Turner 45,X. O fenótipo e o sexo de criação/identidade de gênero são variáveis na MGD, embora o resultado seja frequentemente assimetria gonadal e lábio-escrotal, conforme detalhado na Figura 8. Qualquer paciente com cariótipo 45,X/46,XY pode apresentar manifestações de TS (p.ex., coarctação da aorta, anomalias renais), portanto deve ser submetido aos mesmos exames de rastreamento sistêmico que os pacientes com TS clássica.7,8
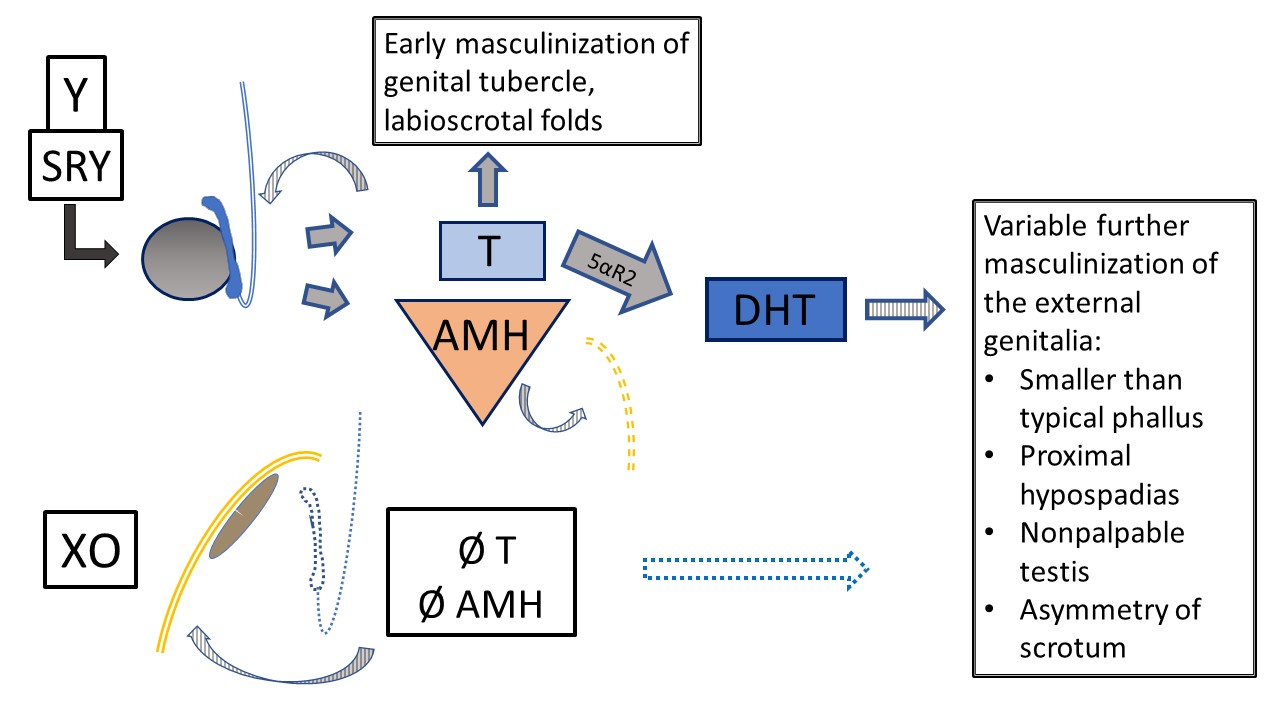
Figura 8 Disgenesia gonadal mista 45,X/46,XY. Vários fenótipos ocorrem com esse cariótipo em mosaico. Este diagrama representa o desenvolvimento interno e externo no contexto de um testículo (determinado pelo cromossomo Y) e uma gônada em fita (desenvolvimento padrão, estroma de tipo ovariano). O fenótipo clássico, quando se observa algum grau de ambiguidade, é: uma gônada não palpável, um testículo descido e hipospádia proximal. Como T e AMH têm função parácrina, a gônada em fita está associada a hemiútero e tuba uterina, sem estruturas de Wolff. Devido ao material do Y, há risco de gonadoblastoma e disgerminoma. O testículo apresenta desenvolvimento disgenético variável, impactando a função precoce e futura e o risco tumoral. Ocorre masculinização primária, mas o desenvolvimento adicional da uretra e do escroto é variavelmente impactado pela função testicular.
DSD ovotesticular
O DSD ovotesticular ocorre quando há tanto tecido ovariano quanto testicular presentes no mesmo indivíduo (antigo nome – hermafroditismo verdadeiro). Tipicamente, o tecido ovariano é mais bem desenvolvido, e o tecido testicular é disgenético. Fenótipo, sexo de criação/identidade de gênero e potencial de fertilidade são todos variáveis. É importante notar que pacientes com DSD ovotesticular podem ter qualquer cariótipo (embora 46, XX seja o mais comum), portanto esse diagnóstico deve ser considerado para qualquer paciente que se apresente com genitália ambígua.
Avaliação Multidisciplinar
Além de propor uma nomenclatura menos pejorativa, outros pilares da declaração de consenso de 2006 são paradigma são o aconselhamento por uma equipe multidisciplinar e o apoio psicossocial longitudinal a pacientes e pais.1
A Equipa Multidisciplinar
A equipe que trabalha com a família em uma consulta durante a internação do recém-nascido é, de preferência, a mesma que trabalhará com pacientes e famílias longitudinalmente. A equipe é responsável por educar a família sobre a condição e todas as potenciais opções de manejo; oferece apoio e envolve a família na tomada de decisão compartilhada quando decisões são necessárias (por exemplo, sexo de criação, divulgação, cirurgia). Idealmente, a equipe inclui os seguintes membros:
- Endocrinologia
- Saúde Mental (Psicólogo, Assistente Social)
- Urologia / Cirurgia Pediátrica / Ginecologia
- Conselheiro Genético / Geneticista
- Enfermagem
- Defensor do paciente / Educador do paciente
- Neonatologista (interconsulta hospitalar)
Cada especialista traz diferentes perspectivas, experiência clínica relevante e habilidades interpessoais para a equipe multidisciplinar. Os cirurgiões são uma parte importante da equipe, embora as necessidades cirúrgicas precoces ou urgentes sejam incomuns. Todos participam das discussões da equipe e contribuem para a experiência do paciente e da família. Idealmente, o respeito mútuo e a disposição para considerar todas as perspectivas aprimoram o funcionamento da equipe e permitem que a equipe ofereça cuidados de nível ainda mais elevado ao longo do tempo, particularmente à medida que os padrões de cuidado evoluem.
Cada indivíduo vivendo com uma condição de DSD pode não ter acesso a uma equipe multidisciplinar funcional. Alguns podem não aderir ao trabalho com a equipe ou podem considerar oneroso o acompanhamento longitudinal. Cada oportunidade precoce de educação sobre o diagnóstico e potenciais preocupações de longo prazo ou necessidades médicas é, portanto, muito importante. Em situações em que o acompanhamento de longo prazo é viável e aceitável, isso pode ser realizado no contexto de um acompanhamento mais regular por uma única especialidade, conforme apropriado para o paciente. Por exemplo, alguns dos pacientes com CAH na instituição dos autores fazem acompanhamento trimestral com seu endocrinologista e participam de uma consulta completa com a equipe multidisciplinar anualmente.
Trabalhar ativamente em conjunto, aprendendo uns com os outros e com pacientes e famílias, facilita o funcionamento da equipa e melhora a assistência. A equipa recorre a experiências prévias e assimila novos conhecimentos, particularmente nos domínios da experiência do paciente e dos testes genéticos de próxima geração. A equipa também identifica lacunas no conhecimento e no paradigma de cuidados que podem ser abordadas por meio de colaborações e investigação direcionada. Conferências multidisciplinares periódicas, baseadas em casos ou em temas, ajudam a construir familiaridade com as várias condições entre os membros da equipa e os formandos e oferecem oportunidade para compreender diferenças de filosofia.
Abordagem Inicial do Paciente com Suspeita de DSD
Pacientes com suspeita de DSD apresentam-se classicamente na infância com genitália ambígua ou, na adolescência, com amenorreia primária. As condições de DSD também podem chegar à atenção médica devido a achados incidentais em exames de imagem ou intraoperatórios, a uma hérnia inguinal em uma menina, ou se a menina apresentar masculinização na época da puberdade. Cada vez mais, as condições de DSD são suspeitadas no período pré-natal devido à discordância entre o DNA livre de células e a ultrassonografia, ou por preocupação com uma diferença no desenvolvimento genital observada na ultrassonografia.
Quando nasce um recém-nascido com suspeita de DSD, recomenda-se a consulta com uma equipe multidisciplinar (descrita acima) sempre que possível. Lembre-se de parabenizar a família pelo novo bebê (uma etapa frequentemente negligenciada, especialmente quando há surpresa ou falta de familiaridade com DSD). Utilize termos neutros quanto ao gênero, (“seu bebê”, não “isso”) ao se referir ao bebê se houver qualquer dúvida quanto à atribuição de sexo. Figura 9 apresenta recomendações para descrever o exame genital do recém-nascido, incluindo termos neutros quanto ao gênero a serem usados. Juntamente com um histórico pré-natal detalhado, histórico familiar e exame físico, as recomendações para a avaliação inicial de um paciente com suspeita de DSD incluem:
- Cariótipo
- Avaliação endócrina (17-OH-progesterona, testosterona, hormônio luteinizante, hormônio folículo-estimulante, eletrólitos)
- Ultrassonografia pélvica para avaliar a posição e o tipo gonadal, e a presença de estruturas müllerianas
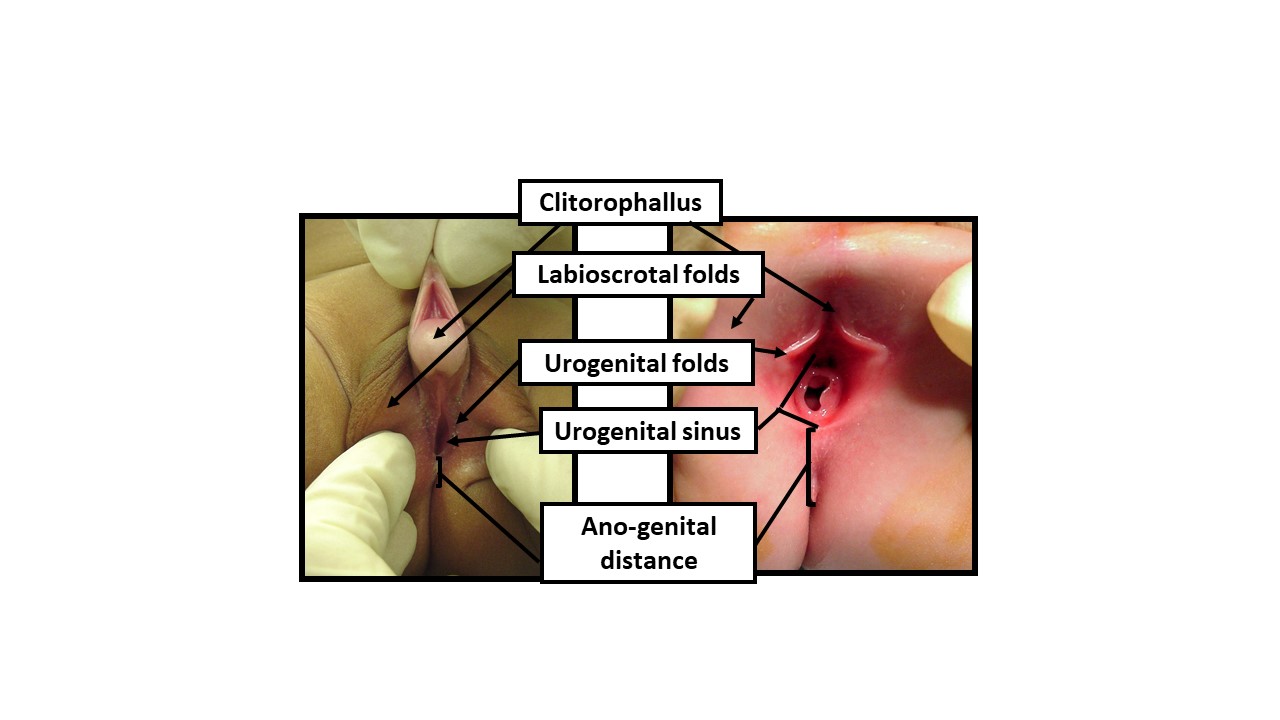
Figura 9 Descrevendo o exame do recém-nascido com termos anatômicos neutros quanto ao gênero. Ao examinar um recém-nascido com aparência ambígua dos genitais, deve-se utilizar terminologia neutra quanto ao gênero até que dados suficientes permitam a tomada de decisão compartilhada com os pais para a determinação do sexo de criação. O exame deve documentar as seguintes manifestações de exposição androgênica: 1) Comprimento da estrutura clitorofálica, 2) Largura da glande, 3) Número e posição dos orifícios perineais, 4) Grau de fusão das pregas labioscrotais, 5) Hiperpigmentação e rugosidade das pregas labioscrotais, 6) Distância anogenital, 7) Localização de quaisquer gônadas palpáveis
Uma abordagem algorítmica baseada no número de gônadas palpáveis e nos resultados endócrinos, genéticos e de exames de imagem, conforme descrito acima, pode ajudar de forma eficiente a restringir o diagnóstico diferencial. No recém-nascido, o mais crítico do ponto de vista médico é excluir a forma perdedora de sal da hiperplasia adrenal congênita (HAC), com risco de vida. Toda a equipe multidisciplinar descrita acima desempenha um papel durante esse período diagnóstico crítico para ajudar a família a integrar novas informações e a tomar decisões sobre sexo de criação, divulgação e cirurgia.
Opções de tratamento cirúrgico
As opções cirúrgicas para pacientes com DSD são altamente individualizadas, e as decisões sobre o tratamento cirúrgico devem, idealmente, ser tomadas no contexto de uma equipe multidisciplinar. Dependendo das necessidades e objetivos do paciente, os procedimentos podem ser diagnósticos, reconstrutivos ou extirpativos.
Procedimentos Diagnósticos
Procedimentos diagnósticos são frequentemente realizados quando a anatomia ou o diagnóstico permanecem pouco claros após avaliação endócrina e genética e exames de imagem radiológica detalhados.
A endoscopia permite visualização detalhada e medições do trato urogenital. Por exemplo, o comprimento e a configuração de um seio urogenital podem ser caracterizados. As estruturas vaginais podem ser avaliadas quanto à profundidade, ao número e à presença ou ausência de colo do útero.
A laparoscopia diagnóstica auxilia na visualização das gônadas e das estruturas müllerianas. Essa visualização pode auxiliar no diagnóstico e fornecer informações para o planejamento cirúrgico reconstrutivo futuro. A biópsia gonadal pode ser realizada por abordagem laparoscópica ou aberta para determinar o tipo de tecido (por exemplo, se houver suspeita de DSD ovotesticular), ou ser enviada para cariotipagem se houver suspeita de material do cromossomo Y que não tenha sido identificado na cariotipagem do sangue periférico. Figura 10 mostra imagens estáticas laparoscópicas de um paciente com MGD: o diagnóstico de MGD foi suspeitado clinicamente, e confirmado pelo aspecto gonadal macroscópico e histológico fornecido pela laparoscopia diagnóstica e pela biópsia.
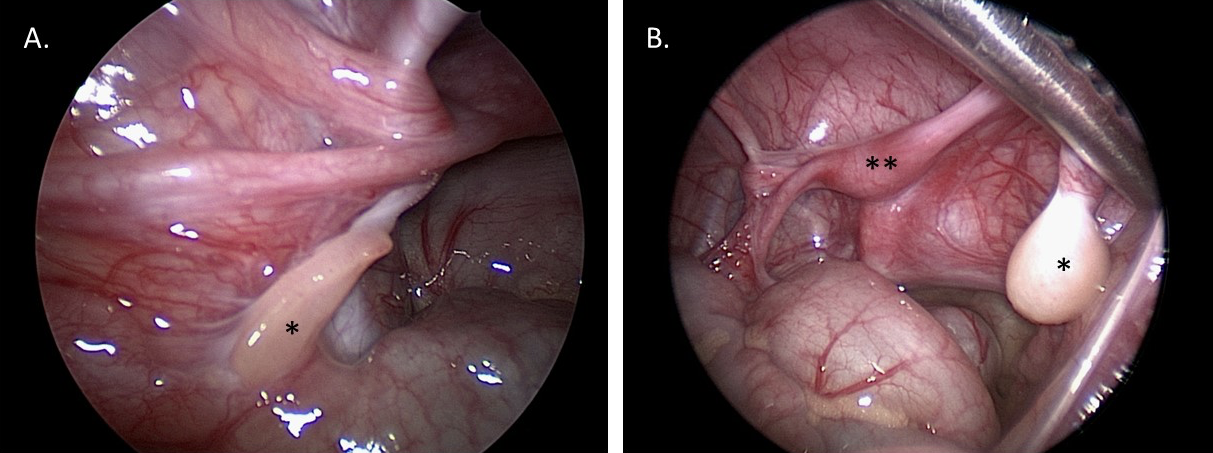
Figura 10 Imagens laparoscópicas diagnósticas da pelve em um paciente de 1 ano de idade com disgenesia gonadal mista. A. *Gônada em fita esquerda. B. *Testículo disgenético direito. **Útero.
Genitoplastia Masculinizante
Para pacientes com DSD, a genitoplastia masculinizante mais frequentemente envolve um reparo de hipospádia proximal, frequentemente com escrotoplastia para corrigir a transposição penoescrotal e/ou uma configuração bífida. Os detalhes técnicos do reparo de hipospádia são semelhantes aos de pacientes sem uma condição de DSD definida. O reparo de hipospádia em pacientes com DSD tipicamente requer um procedimento em dois estágios para obter correção da curvatura peniana e um meato uretral terminal. Há controvérsia na literatura sobre se pacientes com diagnóstico conhecido de DSD apresentam piores desfechos cirúrgicos da hipospádia quando comparados a pacientes com hipospádia proximal que não têm um diagnóstico específico de DSD.9,10,11
Genitoplastia feminizante
Os componentes incluem clitoroplastia (com labioplastia) e vaginoplastia. Existe uma variedade de abordagens, que podem ser adaptadas com base na anatomia e nos objetivos do paciente. Um exemplo de marcações cirúrgicas pré-operatórias para uma paciente com HAC que foi submetida a uma mobilização urogenital parcial (PUM) e a uma labioplastia sem clitoroplastia está ilustrado na Figura 11.
Clitoroplastia e Labioplastia
Os procedimentos modernos de clitoroplastia mais frequentemente relatados envolvem uma redução clitoriana poupadora de nervos.12 O clitóris é desenluvado e o tecido corporal (erétil) é removido, evitando os feixes neurovasculares dorsais. A abordagem poupadora da túnica albugínea na clitoroplastia de redução foi introduzida para preservar ainda mais o suprimento sanguíneo e a sensibilidade da glande e envolve incisões ventrais no tecido corporal e excisão de tecido erétil, preservando a túnica circundante. A glande do clitóris é frequentemente preservada, recuada proximalmente e fixada sob o púbis. A redução da glande pode ser realizada pela excisão de uma cunha ventral de tecido e reaproximando as bordas ventrais entre si. No entanto, essa abordagem traz risco de redução da sensibilidade da glande. Um clitóris oculto pode frequentemente ser obtido com clitoroplastia juntamente com labioplastia, de modo que a redução da glande tem se tornado menos frequente.
Várias técnicas poupadoras dos corpos cavernosos surgiram como alternativas ao procedimento de redução clitoriana descrito acima. Como exemplo, Pippi Salle descreveu um procedimento de desmontagem corporal.13 Nesse procedimento, os corpos cavernosos são completamente separados da glande e do feixe neurovascular, e entre si. Em seguida, os corpos cavernosos são tunelizados individualmente para os respectivos grandes lábios ipsilaterais, e a glande é reposicionada proximalmente, de modo semelhante aos procedimentos de redução clitoriana. Esse ‘procedimento de Pippi Salle’ tem a vantagem teórica de reversibilidade, uma vez que nenhum tecido é removido, embora tentativas de reversão, e outros desfechos em longo prazo, ainda não tenham sido relatados.
A labioplastia é geralmente a etapa final de um procedimento de clitoroplastia (com ou sem vaginoplastia). Os objetivos são reconfigurar o prepúcio clitoriano e o tecido do corpo do clitóris sobre o clitóris para recobrir a glande, e avançar as bordas cutâneas posteriormente, lateralmente ao vestíbulo/abertura vaginal.
Vaginoplastia
A abordagem da vaginoplastia envolve uma avaliação para determinar se a paciente apresenta um seio urogenital com confluência baixa (canal comum curto com separação da vagina e da uretra próxima ao períneo) ou confluência alta (canal comum mais longo e separação mais alta.14
Uma vaginoplastia com retalho cutâneo perineal (técnica de Fortunoff) pode ser suficiente para pacientes com seio urogenital de confluência baixa. Para essa técnica, cria-se um retalho posterior em U (como na Figura 11) A parede posterior da vagina é então incisada proximalmente à confluência displásica, e o retalho posicionado e fixado dentro dessa incisão vaginal para aumentar o calibre vaginal.
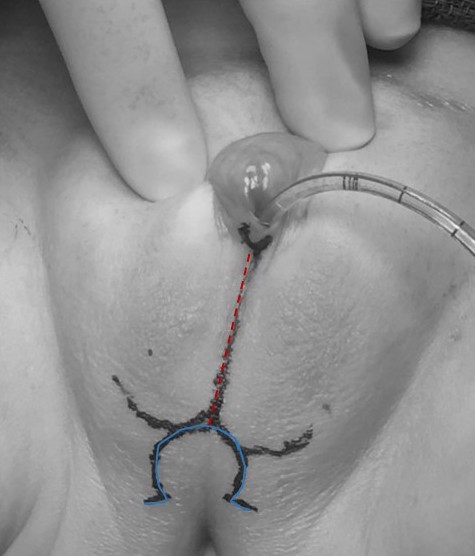
Figura 11 Marcação cirúrgica pré-operatória para uma paciente com hiperplasia adrenal congênita que será submetida a mobilização urogenital parcial e labioplastia sem clitoroplastia. Observa-se um cateter na abertura do seio urogenital. A marcação da linha média (vermelho tracejado) estende-se através das pregas lábio-escrotais fundidas, e a marca em forma de ômega (azul contínuo) formará o retalho cutâneo posterior para a vaginoplastia.
Em mobilização urogenital total (TUM) ou mobilização urogenital parcial (PUM), a uretra e a vagina são mobilizadas em direção ao períneo como uma unidade.15 A distinção entre as duas está em saber se a dissecção anterior atrás do púbis secciona o ligamento pubouretral, como mostrado na Figura 12. Existe a preocupação potencial de incontinência urinária com mobilização mais extensa, mas isso não foi demonstrado de forma definitiva e provavelmente é mais preocupante se a distância da confluência ao colo vesical estiver encurtada. Se aplicada a casos de confluência baixa, a PUM geralmente traz a vagina para uma posição em que pode ser exteriorizada ou ponteada com um retalho perineal de base posterior (ver acima). Confluência alta indica uma anatomia mais complexa, incluindo uma maior distância até o períneo, uma relação mais próxima entre a vagina e o colo vesical e, frequentemente, uma vagina mais curta. O abaixamento direto pode não ser vantajoso a longo prazo. Nos casos de confluência alta, mesmo com mobilização urogenital, será necessário separar a vagina do seio urogenital e abaixá-la até o períneo. Nesse cenário, o seio urogenital torna-se a uretra. A vagina atinge o períneo com o auxílio de retalhos cutâneos perineais ou do uso de tecido excedente do seio urogenital mobilizado como retalho.
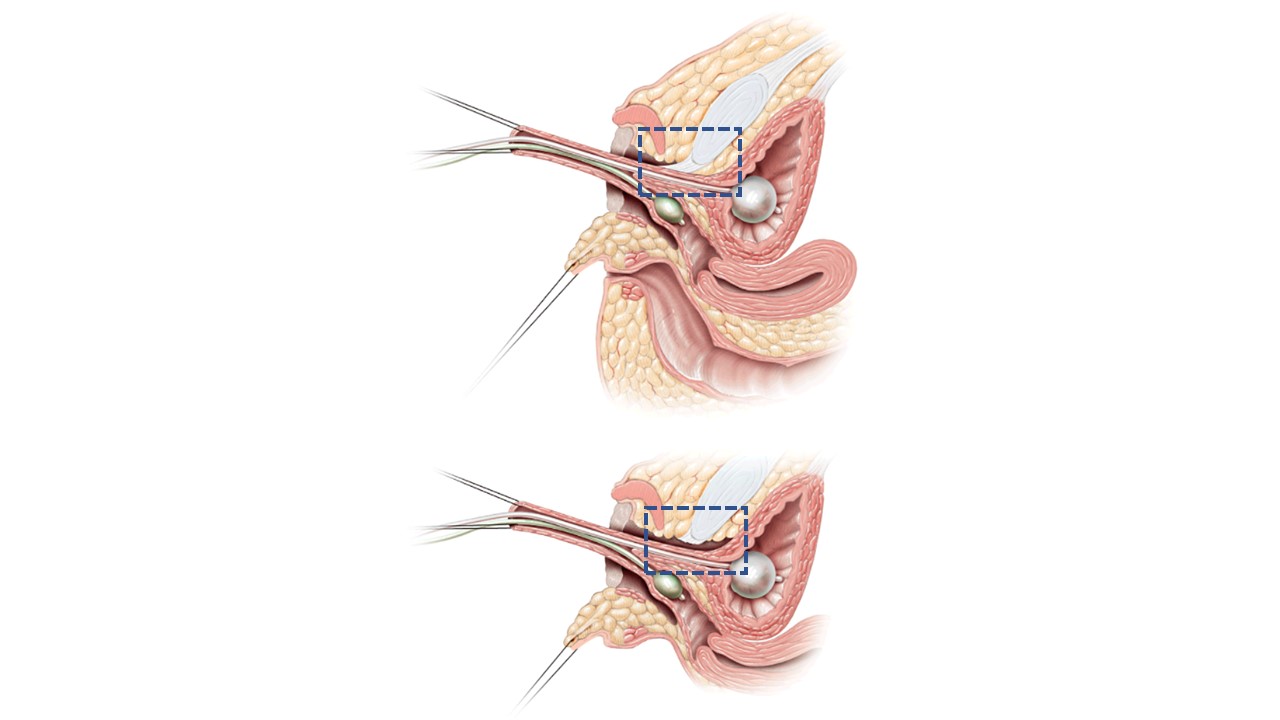
Figura 12 Imagens sagitais em corte transversal da mobilização urogenital parcial vs total. A. Mobilização urogenital parcial: uretra e vagina são avançadas em direção ao períneo como uma unidade. O ligamento pubouretral (caixa de contorno azul tracejado) é mantido intacto. B. Mobilização urogenital total: uretra e vagina são avançadas em direção ao períneo como uma unidade. O ligamento pubouretral (caixa de contorno azul tracejado) é seccionado. Adaptado de: Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int 2008; 102 (9): 1182–1197. DOI: 10.1111/j.1464-410x.2008.08091.x. (permissão concedida em 2/1/2022)
Para pacientes que nascem com estruturas müllerianas ausentes ou pouco desenvolvidas, a vaginoplastia envolve a criação romba de uma neovagina no espaço retrovesical entre a bexiga e o reto, e o revestimento do novo espaço potencial com tecido autólogo (por exemplo, enxerto de pele, enxerto de mucosa bucal, retalho peritoneal) ou outros enxertos biológicos (por exemplo, submucosa do intestino delgado [SIS]). Essas técnicas também podem ser usadas como adjuvantes aos procedimentos descritos acima, ou no cenário de reoperação.
Dilatações vaginais
Alguns pacientes com DSD (por exemplo, SIA completa) podem alcançar, com dilatações vaginais regulares, uma vagina que permita atividade sexual confortável e prazerosa, sem precisar de uma vaginoplastia cirúrgica. Todos os pacientes que se submetem a uma vaginoplastia cirúrgica também necessitam de dilatações vaginais durante a cicatrização após a cirurgia e até que estejam tendo relações sexuais vaginais de maneira regular.
Gonadectomia e Manejo Gonadal
O paradigma do manejo gonadal em pacientes com DSD evoluiu ao longo do tempo. Tradicionalmente, pacientes com DSD em que há risco aumentado de formação tumoral eram orientados a realizar gonadectomia. Presumia-se infertilidade. Com o tempo, tornou-se evidente que há uma ampla variação do risco tumoral e que os tumores que se formam são frequentemente gonadoblastomas (não malignos).16,17 Disgerminomas (tumores malignos de células germinativas) são possíveis, mas extremamente raros antes da puberdade. Também pode haver potencial de fertilidade não tradicional para alguns que anteriormente eram considerados inférteis.18 Assim, as recomendações de manejo gonadal estão se tornando mais individualizadas, considerando o diagnóstico do paciente (e, portanto, o risco tumoral previsto), a idade e o potencial para função gonadal endógena (tanto hormonal quanto reprodutiva). Para pacientes que optam por gonadectomia, a criopreservação de tecido gonadal é uma opção emergente que pode permitir fertilidade biológica, assumindo que ocorram os avanços futuros esperados nas tecnologias de reprodução assistida.19 Ainda não há protocolos de vigilância baseados em evidências para pacientes que optam por manter as gônadas; a instituição dos autores normalmente recomenda ultrassonografia pélvica a cada 6–12 meses. Tabela 4 apresenta estratégias de recomendação gonadal para vários diagnósticos de DSD.
Tabela 4 Opções de manejo gonadal por diagnóstico. * considere a criopreservação experimental de tecido gonadal.
| Diagnóstico | Risco Tumoral Estimado | Células Germinativas Esperadas? | Puberdade Esperada? | Opções de Manejo das Gônadas |
|---|---|---|---|---|
| 46, XY Disgenesia Gonadal Completa (Síndrome de Swyer) | Alto – Até 35% |
Não (talvez células germinativas precursoras) |
Não | -Gonadectomia* |
| Síndrome de Insensibilidade Parcial aos Andrógenos | Alto – Até 50% dependendo da localização da gônada |
Sim Espermatozoides possíveis |
Sim, variável | -Orquidopexia, considerar biópsia -Observação c/ ultrassonografias -Gonadectomia* |
| Síndrome de Turner + Cromossomo Y | Médio – ~10-30% |
Talvez – Óvulos possíveis | Possível | -Observação c/ultrassonografias -Gonadectomia* (padrão, baixo nível de evidência) |
| Síndrome de Insensibilidade Completa aos Andrógenos | Baixo – ~2-10% |
Sim – Precursores de espermatozoides |
Sim – devido à aromatização da testosterona em estrogênio | -Gonadectomia pós-puberal* -Observação c/ultrassonografias, RM |
Manejo das Estruturas Müllerianas
Para pacientes que estão sendo criados como do sexo masculino e têm estruturas müllerianas persistentes (isto é, um utrículo grande), estas geralmente são mantidas in situ, a menos que se tornem sintomáticas. A excisão implica risco de dano potencial às estruturas dos vasos deferentes (e possivelmente aos nervos pélvicos), e os pacientes são frequentemente assintomáticos. A correção da hipospádia é segura, e a excisão mesmo de grandes estruturas utriculares não é obrigatória antes da correção da hipospádia. Se ocorrerem sintomas como infecções ou hematúria cíclica incômoda, pode-se realizar excisão cirúrgica por via aberta, laparoscópica ou robótica. O tratamento hormonal com supressão menstrual também pode ser considerado para pacientes que apresentam dor cíclica devido a um remanescente mülleriano obstruído, ou hematúria cíclica incômoda.
Conclusões
Os DSD abrangem uma ampla variedade de condições que resultam de desenvolvimento cromossômico, gonadal ou anatômico anormal. Existe uma ampla variedade de fenótipos, mesmo dentro de diagnósticos e categorias específicas de DSD. Recomenda-se, sempre que possível, uma abordagem multidisciplinar para avaliação e acompanhamento a longo prazo.
Pontos-chave
- A terminologia de DSD aceita pela medicina não é a preferida por todos os indivíduos afetados e suas famílias. Em encontros clínicos individuais, pergunte aos pacientes e às famílias quais termos preferem usar ao se referirem ao diagnóstico e à anatomia.
- As condições de DSD ocorrem devido a cromossomos, gônadas ou ação hormonal atípicos. Refletir sobre o grau de anormalidade e determinar se o defeito identificado é completo ou parcial pode ajudar a prever o fenótipo do paciente.
- O número de gônadas palpáveis estabelece a base para um diagnóstico diferencial de DSD. O diferencial é posteriormente refinado por meio de cariótipo, exames hormonais e ultrassonografia pélvica.
- Lembre-se de parabenizar os novos pais que têm um bebê com suspeita de DSD—essa etapa crítica é frequentemente esquecida.
- CAH é a forma mais comum de DSD 46, XX. As características clínicas incluem genitália externa masculinizada com gônadas não palpáveis. Perda salina com risco de vida pode ocorrer na 1ª semana de vida.
- DSD ovotesticular pode ocorrer em um paciente com qualquer cariótipo.
- Um paciente com cariótipo 45, X/46, XY pode apresentar genitália externa típica feminina, ambígua ou típica masculina. Qualquer paciente com esse cariótipo pode apresentar manifestações sistêmicas da Síndrome de Turner.
Leituras recomendadas
- Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
- Kaefer M, Diamond D, Hendren WH. The Incidence Of Intersexuality In Children With Cryptorchidism And Hypospadias: Stratification Based On Gonadal Palpability And Meatal Position. J Urol 1999; 162 (3 Part 2): 1006–1007. DOI: 10.1016/s0022-5347(01)68049-2.
- Kim S, Rosoklija I, Johnson EK. Surgical, Patient, and Parental Considerations in the Management of Children with Differences of Sex Development. Curr Pediatr Rep 2018; 6 (3): 209–219. DOI: 10.1007/s40124-018-0177-4.
- Mouriquand PD, Gorduza DB, Gay CL. Re: “Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how?” J Pediatr Urol 2016; 12 (6): 442–443. DOI: 10.1016/j.jpurol.2016.07.013.
- Nixon R, Cerqueira V, Kyriakou A, Lucas-Herald A, McNeilly J, McMillan M, et al.. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod 2017; 32 (10): 2130–2137. DOI: 10.1093/humrep/dex280.
- Van Batavia JP, Kolon TF. Fertility in disorders of sex development: A review. J Pediatr Urol 2016; 12 (6): 418–425. DOI: 10.1016/j.jpurol.2016.09.015.
Referências
- Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
- Johnson EK, Rosoklija I, Finlayson C. Faculty Opinions recommendation of Attitudes towards "disorders of sex development" nomenclature among affected individuals. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2017; 13: 608. DOI: 10.3410/f.727655647.793533819.
- D’Oro A, Rosoklija I, Jacobson DL, Finlayson C, Chen D, Tu DD, et al.. Patient and Caregiver Attitudes toward Disorders of Sex Development Nomenclature. J Urol 2020; 204 (4): 835–842. DOI: 10.1097/ju.0000000000001076.
- Davies JH, Knight EJ, Savage A, Brown J, Malone PS. Evaluation of terminology used to describe disorders of sex development. J Pediatr Urol 2011; 7 (4): 412–415. DOI: 10.1016/j.jpurol.2010.07.004.
- Lin-Su K, Lekarev O, Poppas DP, Vogiatzi MG. Congenital adrenal hyperplasia patient perception of ‘disorders of sex development’ nomenclature. Int J Pediatr Endocrinol 2015; 2015 (1): 9. DOI: 10.1186/s13633-015-0004-4.
- Snodgrass W, Macedo A, Hoebeke P, Mouriquand PDE. Hypospadias dilemmas: A round table. J Pediatr Urol 2011; 7 (2): 145–157. DOI: 10.1016/j.jpurol.2010.11.009.
- Wu Q, Wang C, Shi H, Kong X, Ren S, Jiang M. The Clinical Manifestation and Genetic Evaluation in Patients with 45,X/46,XY Mosaicism. Sex Dev 2017; 11 (2): 64–69. DOI: 10.1159/000455260.
- Das DV, Jabbar PK. Clinical and Reproductive Characteristics of Patients with Mixed Gonadal Dysgenesis (45,X/46, XY). J Obstet Gynaecol India 2021; 71 (4): 399–405. DOI: 10.1007/s13224-021-01448-3.
- Saltzman AF, Carrasco A, Colvin A, Campbell JB, Vemulakonda VM, Wilcox D. Patients with disorders of sex development and proximal hypospadias are at high risk for reoperation. World J Urol 2018; 36 (12): 2051–2058. DOI: 10.1007/s00345-018-2350-3.
- Ochi T, Ishiyama A, Yazaki Y, Murakami H, Takeda M, Seo S, et al.. Surgical management of hypospadias in cases with concomitant disorders of sex development. Pediatr Surg Int 2019; 35 (5): 611–617. DOI: 10.1007/s00383-019-04457-6.
- Palmer BW, Reiner W, Kropp BP. Proximal Hypospadias Repair Outcomes in Patients with a Specific Disorder of Sexual Development Diagnosis. Adv Urol 2012; 2012: 1–4. DOI: 10.1155/2012/708301.
- Kaefer M, Rink RC. Treatment of the Enlarged Clitoris. Front Pediatr 2017; 5. DOI: 10.3389/fped.2017.00125.
- Pippi Salle JL, Braga LP, Macedo N, Rosito N, Bagli D. Corporeal Sparing Dismembered Clitoroplasty: An Alternative Technique for Feminizing Genitoplasty. J Urol 2007; 178 (4s): 1796–1801. DOI: 10.1016/j.juro.2007.03.167.
- Guarino N, Scommegna S, Majore S, Rapone AM, Ungaro L, Morrone A, et al.. Vaginoplasty for Disorders of Sex Development. Front Endocrinol (Lausanne) 2013; 4: 29, DOI: 10.3389/fendo.2013.00029.
- Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int 2008; 102 (9): 1182–1197. DOI: 10.1111/j.1464-410x.2008.08091.x.
- Looijenga LHJ, Hersmus R, Oosterhuis JW, Cools M, Drop SLS, Wolffenbuttel KP. Tumor risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab 2007; 21 (3): 480–495. DOI: 10.1016/j.beem.2007.05.001.
- Looijenga LHJ, Hersmus R, Leeuw BHCGM de, Stoop H, Cools M, Oosterhuis JW, et al.. Gonadal tumours and DSD. Best Pract Res Clin Endocrinol Metab 2010; 24 (2): 291–310. DOI: 10.1016/j.beem.2009.10.002.
- Finlayson C, Fritsch MK, Johnson EK, Rosoklija I, Gosiengfiao Y, Yerkes E, et al.. Presence of Germ Cells in Disorders of Sex Development: Implications for Fertility Potential and Preservation. J Urol 2017; 197 (3 Part 2): 937–943. DOI: 10.1016/j.juro.2016.08.108.
- Harris CJ, Corkum KS, Finlayson C, Rowell EE, Laronda MM, Reimann MB, et al.. Establishing an Institutional Gonadal Tissue Cryopreservation Protocol for Patients with Differences of Sex Development. J Urol 2020; 204 (5): 1054–1061. DOI: 10.1097/ju.0000000000001128.
- Kaefer M, Diamond D, Hendren WH. The Incidence Of Intersexuality In Children With Cryptorchidism And Hypospadias: Stratification Based On Gonadal Palpability And Meatal Position. J Urol 1999; 162 (3 Part 2): 1006–1007. DOI: 10.1016/s0022-5347(01)68049-2.
- Kim S, Rosoklija I, Johnson EK. Surgical, Patient, and Parental Considerations in the Management of Children with Differences of Sex Development. Curr Pediatr Rep 2018; 6 (3): 209–219. DOI: 10.1007/s40124-018-0177-4.
- Mouriquand PD, Gorduza DB, Gay CL. Re: “Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how?” J Pediatr Urol 2016; 12 (6): 442–443. DOI: 10.1016/j.jpurol.2016.07.013.
- Nixon R, Cerqueira V, Kyriakou A, Lucas-Herald A, McNeilly J, McMillan M, et al.. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod 2017; 32 (10): 2130–2137. DOI: 10.1093/humrep/dex280.
- Van Batavia JP, Kolon TF. Fertility in disorders of sex development: A review. J Pediatr Urol 2016; 12 (6): 418–425. DOI: 10.1016/j.jpurol.2016.09.015.
Ultima atualização: 2025-09-21 13:35