
13: Doenças Císticas do Rim
Este capítulo levará aproximadamente 23 minutos para ler.
Introdução
Doenças císticas renais constituem uma ampla categoria de condições renais esporádicas e genéticas, congênitas ou adquiridas, que podem envolver a presença de um único cisto a muitos cistos em um ou ambos os rins (ver classificações hereditárias na Tabela 1 abaixo).1 Com base em um Comitê de 1987 sobre Classificação, Nomenclatura e Terminologia, a classificação da doença cística renal é determinada principalmente pela etiologia genética ou não genética.2 As doenças císticas renais podem, em grande parte, ser consideradas condições raras; no entanto, podem estar associadas a consequências graves, como doença renal crônica (DRC), insuficiência renal ou doença hepática. Apesar de sua raridade, a doença renal policística autossômica dominante (DRPAD) é uma das condições hereditárias humanas mais comuns. A possibilidade de complicações graves decorrentes das doenças císticas renais na população pediátrica, especialmente DRC, torna o diagnóstico precoce e modalidades terapêuticas eficazes de particular importância.
Tabela 1 Classificações hereditárias das doenças renais císticas.1
| Hereditárias | Não hereditárias |
|---|---|
| Doença renal policística autossômica recessiva (infantil) | Rim multicístico (rim displásico multicístico) |
| Doença renal policística autossômica dominante (do adulto) | Cisto multilocular benigno (nefroma cístico) |
| Complexo nefronoftise juvenil e doença cística medular | Cistos simples |
| Nefronoftise juvenil (autossômica recessiva) | Rim em esponja medular |
| Doença cística medular (autossômica dominante) | Doença renal glomerulocística esporádica |
| Nefrose congênita (síndrome nefrótica familiar) (autossômica recessiva) | Doença renal cística adquirida |
| Doença glomerulocística hipoplásica familiar (autossômica dominante) | Divertículo calicinal (cisto pielogênico) |
| Síndromes de malformação múltipla com cistos renais (p.ex., esclerose tuberosa, doença de von Hippel-Lindau) |
Embriologia
Cistos tipicamente ocorrem secundariamente a desequilíbrios secretores, absortivos e eletrolíticos nas células epiteliais e se formam em qualquer local do sistema tubular renal. Pesquisas recentes sobre doença renal cística hereditária sugerem que os genes afetados envolvem o cílio primário das células epiteliais renais, uma via comum potencial para essas condições.3 O termo “ciliopatia” refere-se a essa via comum. Os cílios têm a função de modular a proliferação e a diferenciação das células tubulares, e sua disfunção pode levar a uma expansão inadequada dos túbulos e à formação de cistos.4
Este capítulo discutirá a epidemiologia, a patogênese, a avaliação e o tratamento de várias doenças renais císticas hereditárias e não hereditárias, incluindo ADPKD, doença renal policística autossômica recessiva (ARPKD), cistos renais isolados, divertículos caliciais, rim displásico multicístico (MCDK), rim em esponja medular, nefronoftise, cistos multiloculares (nefroma cístico multilocular), doenças renais císticas adquiridas e síndromes associadas à doença renal cística.
Doença Renal Policística Autossômica Dominante
A DRPAD é relativamente comum, com incidência de 1 em 400 a 1.000 nascidos vivos, manifestando-se como dilatação cística do néfron e outras anomalias extrarrenais, como aneurisma intracraniano.5 A gravidade varia entre os pacientes e tende a piorar com o tempo; assim, muitos pacientes pediátricos e até alguns adultos de meia-idade podem ser assintomáticos. Embora incomum, doença grave na infância pode acarretar morbidade e mortalidade significativas.6 Devido à história familiar ou a achados incidentais em exames de imagem, mais crianças assintomáticas estão sendo identificadas com DRPAD.
Epidemiologia
Entre os indivíduos acometidos pela DRPAD, cerca de 96% apresentarão sinais clínicos da doença até os 90 anos de idade.7 Apesar de a maioria dos casos ser descoberta em pacientes de meia-idade (30–40 anos) e de a insuficiência renal antes dos 40 anos de idade ser rara, a doença já foi descrita tão cedo quanto no período neonatal. Quando descrita em recém-nascidos, a DRPAD é considerada mais agressiva.8 Apesar do padrão de herança autossômica dominante, com penetrância teórica de 100%, 10% dos casos podem ocorrer esporadicamente.1
Patogênese
A ADPKD é herdada de forma autossômica dominante e as principais mutações genéticas ocorrem em PKD1 e PKD2 nos cromossomos 16 e 4, respectivamente.5 O produto gênico é uma proteína transmembrana (policistina 1 e policistina 2, respectivamente) localizada nos cílios primários.9 As mutações em PKD1 no cromossomo 16 correspondem à maioria dos casos e a maior gravidade, enquanto a maioria dos casos restantes decorre de mutações em PKD2.10
O funcionamento inadequado da policistina 1 ou da policistina 2 leva à desregulação de sinais das vias proliferativas, como cAMP, ERK e mTOR. Os cílios podem desempenhar um papel organizador na transdução desses sinais, pois a desregulação leva à formação de cistos.11
Avaliação e Diagnóstico
Como um exame de imagem custo-efetivo e minimamente invasivo, a ultrassonografia renal é frequentemente utilizada para identificar DRPAD. Os critérios diagnósticos ultrassonográficos em indivíduos com menos de 30 anos incluem a presença de pelo menos dois cistos unilaterais ou bilaterais, sendo necessários mais cistos para sugerir o diagnóstico à medida que a idade aumenta.12 No entanto, DRPAD e DRPAR podem ser difíceis de distinguir na população pediátrica, tornando a presença de história familiar positiva crucial no diagnóstico.13 Na ausência de história familiar, podem ser realizados testes genéticos. Outros fatores importantes a considerar, quando a história familiar é negativa, incluem aumento renal bilateral, três ou mais cistos hepáticos, aneurisma de artéria cerebral e cistos solitários de outros órgãos (aracnoide, glândula pineal, pâncreas ou baço).1,14 Exames de imagem ou biópsia do fígado também podem ajudar a diferenciar DRPAD e DRPAR, pois a fibrose hepática está sempre presente na DRPAR, mas é rara na DRPAD.5 Por fim, a patologia renal (biópsia) pode ser útil, já que a DRPAD pode envolver a totalidade do túbulo, incluindo o glomérulo; no entanto, a DRPAR não apresentaria cistos glomerulares.5
Opções de Tratamento, Desfechos e Complicações
Atualmente, não há cura para a ADPKD. Os objetivos do tratamento incluem retardar o início da doença renal em estágio terminal (ESRD) e reduzir a carga de complicações da ADPKD relacionadas ao declínio da função renal, à doença cardíaca e à hemorragia intracraniana. As complicações são mais significativamente moduladas pelo manejo da hipertensão arterial.1 Portanto, recomenda-se o controle rigoroso da pressão arterial, e os inibidores da enzima conversora da angiotensina (ACE) e os bloqueadores dos receptores de angiotensina são uma escolha comum; no entanto, não há consenso sobre os anti-hipertensivos ideais.15,16
Terapias emergentes sob investigação visam limitar o crescimento de cistos ao reduzir o AMPc com o uso de inibidores de mTOR, somatostatina, inibidores da quinase de tirosina, inibidores da quinase Src, inibidores de MEK e inibidores das quinases dependentes de ciclina.
Doença Renal Poliquística Autossômica Recessiva
A ARPKD é caracterizada por aumento rápido de ambos os rins em lactentes devido à formação de cistos no sistema de ductos coletores. Pode estar associada à fibrose hepática congênita, que pode levar à hipertensão portal. Uma apresentação mais precoce da ARPKD geralmente está associada a maior gravidade.1 Seus efeitos multissistêmicos podem exigir manejo multidisciplinar.
Epidemiologia
Como a ARPKD é menos comum do que a ADPKD, há menos dados epidemiológicos. A incidência descrita é de 1 em 26.500 nascidos vivos, mas pode variar de 1 em 10.000 a 1 em 50.000 nascidos vivos.1,17 No entanto, pode ser mais comum em populações isoladas ou consanguíneas.
Patogénese
ARPKD é herdada em um padrão classicamente autossômico recessivo, com pais heterozigotos não afetados apresentando 25% de risco de ter um filho afetado. É causada por mutação do gene da doença renal e hepática policística 1 (PKHD1) no cromossomo 6, que codifica a fibrocistina (também conhecida como poliductina), uma proteína identificada no sistema tubular renal (ducto coletor e ramo ascendente espesso), em células epiteliais do ducto biliar hepático e em cílios apicais primários.5,18,19
Um paciente típico pode apresentar-se no período neonatal com história de oligoidrâmnio pré-natal, rins aumentados e sequência de “Potter” com hipoplasia pulmonar e óbito perinatal em aproximadamente 30% dos recém-nascidos afetados.20,21 Rins aumentados apresentam-se ecogênicos com dilatação fusiforme dos ductos coletores, e a progressão para doença renal em estágio terminal pode ocorrer em idades variadas. O efeito sobre outros sistemas orgânicos pode manifestar-se como ductos biliares dilatados, fibrose hepática congênita, hipertensão portal e disfunção neurocognitiva.22
Avaliação e Diagnóstico
A ultrassonografia fetal e do recém-nascido pode mostrar rins com acometimento bilateral, muito aumentados e difusamente ecogênicos devido a abundantes microcistos.1 A presença de macrocistos (> 10 mm) pode indicar outras ciliopatias, como MCDK ou ADPKD, e a RM pode ajudar a avaliar melhor as anomalias no contexto de oligoidrâmnio grave.13 O diagnóstico preciso depende de achados de imagem sugestivos, presença de história familiar com modo de herança recessivo, biópsia hepática mostrando lesões periportais (presentes na ARPKD e raras na ADPKD) e ausência de manifestações extrarrenais associadas a outras doenças renais císticas.1
Opções de tratamento, desfechos e complicações
Semelhante à ADPKD, não há cura para ARPKD; assim, muitas opções de tratamento destinam-se ao controle de sintomas ou à paliação relacionada à hipertensão, insuficiência cardíaca congestiva e insuficiências renal e hepática. Tratamentos agressivos, como nefrectomia unilateral ou bilateral, podem ser indicados em indivíduos gravemente afetados que apresentem comprometimento respiratório ou nutricional devido ao efeito de massa de rins aumentados. Terapias descompressivas podem ser implementadas para o manejo da hipertensão portal. Hemodiálise e transplante renal são frequentemente considerados.1
Cistos Renais Isolados
Cistos renais solitários e múltiplos são as lesões mais comuns do rim em adultos e idosos, mas são raros em crianças e lactentes.4 Eles têm sido identificados no período pré-natal, embora as lesões pré-natais comumente se resolvam durante a gravidez.23
Epidemiologia
A ultrassonografia pré-natal mostrou que a prevalência de cistos renais é de 0,09%.23 A incidência de cistos simples do nascimento até os 18 anos de idade é, em média, de 0,22%, com intervalo de 0,1% a 0,45%.24 A incidência aumenta em adultos e com o avançar da idade.
Patogênese
Há grande variedade de tamanhos; no entanto, a maioria permanece com menos de 2 cm e é frequentemente cortical, alterando o contorno renal, mas também pode ser cortical profunda ou aparentemente medular, porém sem continuidade com a pelve renal.1 Tipicamente, não comprometem a função renal, mas podem ocasionalmente causar dor quando o cisto comprime estruturas adjacentes ou obstrui o sistema coletor.5
Avaliação e Diagnóstico
A identificação geralmente é incidental e o diagnóstico pode ser estabelecido com segurança por ultrassonografia quando atendidos os seguintes critérios: 1) cisto internamente anecóico, 2) parede fina, bem delimitada, com margens nítidas, 3) transmissão adequada das ondas de ultrassom com reforço acústico posterior ao cisto, 4) formato esférico ou ovoide.1 Se esses critérios não forem atendidos ou quando a ultrassonografia não fornecer avaliação satisfatória, como no caso de cistos mais complexos, a tomografia computadorizada pode fornecer mais detalhes anatômicos. Alternativamente, a ressonância magnética ou a aspiração por agulha podem ser indicadas.25
Cistos simples não se comunicam com a pelve renal, porém alguns cistos podem aparecer em estreita proximidade. A suspeita de comunicação pode ser avaliada por imagem de corte transversal com contraste intravenoso e fase tardia, como urograma por TC ou urograma por RM. Além disso, a aspiração do cisto e exames laboratoriais podem ajudar a identificar comunicação com a pelve renal. Se não contíguo, o nitrogênio ureico sanguíneo e a creatinina serão comparáveis aos valores do paciente.5,26,27 Um cisto único também pode levantar preocupação para ADPKD no contexto de história familiar positiva.28 Historicamente, a classificação de cistos de Bosniak não foi bem validada em crianças; no entanto, um estudo recente conclui que um sistema Bosniak modificado fornece estratificação de risco razoável e mostrou que lesões com escore mBosniak 1 ou 2 são frequentemente benignas, enquanto escores 3 ou 4 demonstraram ter 90% contendo patologia intermediária ou maligna.29
Opções de tratamento e seus desfechos
Cistos sintomáticos podem ser drenados, porém é provável que recorram sem o uso de um agente esclerosante.5 Outra opção de manejo inclui intervenção cirúrgica com decorticação do cisto, que é passível de abordagem minimamente invasiva, laparoscópica, especialmente para cistos relativamente superficiais e/ou exofíticos.
O seguimento pode consistir na realização de ultrassonografia para monitorar sinais sugestivos de malignidade. Cistos estáveis que não aumentaram de tamanho por dois anos podem não requerer seguimento adicional.29,30,31,32,33 A intervenção pode não estar indicada após comprovação de que a lesão não é maligna.
Divertículos caliciais
Divertículos caliciais (DC) são evaginações raras dos cálices para o parênquima renal. Classificam-se em dois grupos: os do tipo 1 são mais comuns e adjacentes a um cálice superior ou inferior, enquanto os do tipo 2 são maiores, comunicam-se com a pelve renal e são mais frequentemente sintomáticos.34
Epidemiologia
Relatou-se a presença de CD em 0,2% a 0,6% das crianças que foram submetidas a urogramas intravenosos.13
Patogênese
A patogênese da CD é amplamente desconhecida e não é considerada uma doença herdada geneticamente. No entanto, as teorias incluem mutações genéticas que induzem síndromes que afetam o rim ou, alternativamente, falha da regressão do broto ureteral ou causas obstrutivas.13 O cisto é revestido por epitélio de células transicionais e se comunica com o sistema coletor, tipicamente através de um cálice ou de um infundíbulo estreitado ou estenosado e, menos frequentemente, através da pelve renal.35
Avaliação e Diagnóstico
Os pacientes podem ser assintomáticos ou, ao contrário, apresentar hematúria, dor, ITU ou nefrolitíase; o sinal de apresentação mais comum em crianças é ITU febril.35 Divertículos assintomáticos são tipicamente descobertos em estudos de imagem. Embora a ultrassonografia possa identificar inicialmente o divertículo calicial, para distinguir entre um cisto simples e um divertículo calicial, o estudo de imagem mais preciso é a imagem seccional com contraste intravenoso (IV) e fase tardia.35,36,37 Figura 1 e Figura 2 demonstram achados compatíveis com CD na urografia por RM. Um estudo recente que comparou ultrassonografia com imagem seccional constatou que a ultrassonografia renal direcionada teve sensibilidade de 40% e especificidade de 100%, respectivamente, enquanto a imagem seccional (especificamente a urografia por RM com técnica de fluido estático) teve sensibilidade de 100% e especificidade de 91,6%, respectivamente.37 Dor, infecção, formação de abscesso, urosepsia e cálculos renais sintomáticos podem ser todas indicações possíveis para tratamento cirúrgico.
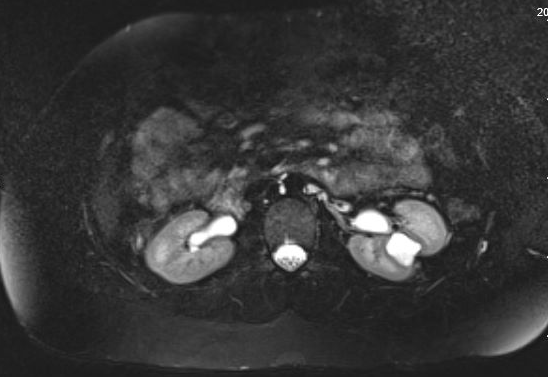
Figura 1 Urograma por Ressonância Magnética mostrando preenchimento por contraste de um divertículo calicial na região interpolar do rim esquerdo, com adelgaçamento sobrejacente do parênquima renal.
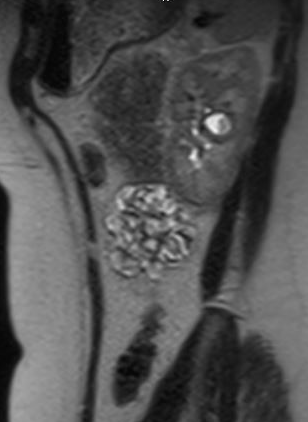
Figura 2 Vista sagital da urografia por RM no mesmo paciente, mostrando contraste preenchendo o divertículo calicial.
Opções de tratamento, seus desfechos e complicações
Pacientes assintomáticos não requerem tratamento, mas podem se beneficiar de monitorização ultrassonográfica.1 Aqueles com indicações para manejo cirúrgico, conforme mencionado acima, podem se beneficiar de marsupialização laparoscópica do divertículo com fulguração do revestimento epitelial.38,39,40 Outros possíveis procedimentos de manejo podem incluir ureteroscopia ou nefrolitotomia percutânea minimamente invasiva (no contexto de cálculos),41 ablação ureteroscópica da cavidade,39 ou ablação e fulguração percutâneas.42 Apesar da variedade de possibilidades terapêuticas, a intervenção cirúrgica para manejar CD pode ser desafiadora. Na tentativa de minimizar a morbidade, o manejo tipicamente começa pelas opções menos invasivas, como abordagens endoscópicas ou percutâneas. Pode haver alta taxa de recorrência de sintomas,42 caso em que a intervenção pode escalar para abordagens laparoscópicas/robóticas e até mesmo abertas, incluindo nefrectomia parcial ou nefrectomia total. Portanto, é necessário aconselhamento extensivo dos pacientes ao considerar intervenção cirúrgica para CD.
Rim displásico multicístico
O rim multicístico displásico (MCDK) é uma malformação congênita comumente diagnosticada na infância ou por ultrassonografia pré-natal, caracterizada por numerosos cistos unilaterais não comunicantes e diferenciação metanéfrica anormal (presença de cartilagem, mesênquima indiferenciado e ductos coletores imaturos), frequentemente no contexto de uropatia obstrutiva.4,13,43 A forma bilateral é geralmente incompatível com a vida devido a oligodrâmnio ou anidrâmnio, levando à hipoplasia pulmonar.44
Epidemiologia
MCDK é uma das anomalias mais comuns, com incidência estimada de 1 em 1.000 a 4.000 nascimentos.1,45 Uma incidência mais elevada de rim único na idade adulta provavelmente está associada à involução do MCDK ao longo do tempo, dada a baixa incidência de agenesia renal verdadeira.5
Patogênese
Displasia multiquística surge antes da formação do néfron, resultante de diferenciação metanéfrica anormal, anomalia do blastema nefrogênico ou obstrução de alto grau ocorrendo durante o desenvolvimento renal.1 Os cistos são revestidos por epitélio cúbico baixo, circundado por células fusiformes, e podem estar preenchidos por fluido proteináceo ou sanguinolento.1 Componentes primitivos e displásicos, assim como cartilagem imatura ou glomérulos imaturos, podem ser observados. A MCDK é classificada como tipo infundibulopélvico, com atresia da pelve renal e do ureter, versus o tipo hidronefrótico, menos comum, que envolve apenas atresia do ureter proximal.46 O rim contralateral pode apresentar hipertrofia compensatória.
Avaliação e Diagnóstico
Atualmente, o MCDK é mais frequentemente identificado na ultrassonografia pré-natal, enquanto a avaliação de uma massa palpável em um neonato é a segunda apresentação mais comum.47
O diagnóstico de MCDK deve ser diferenciado de hidronefrose decorrente de outras condições, como obstrução da junção ureteropélvica. Notadamente, em MCDK, os cistos não se comunicam e cistos maiores aparecem lateralmente. Na hidronefrose, o parênquima renal circunda a estrutura cística central e a típica forma reniforme é mantida.48 Figura 3 e Figura 4 ajudam a demonstrar as dificuldades de diferenciar MCDK de hidronefrose na ultrassonografia. Figura 5 ilustra cistos não comunicantes de vários tamanhos, típicos de MCDK. Para esclarecer melhor, pode ser realizada uma cintilografia com radionuclídeos, que mostraria captação na hidronefrose e, tipicamente, captação mínima ou ausente no contexto de MCDK.49
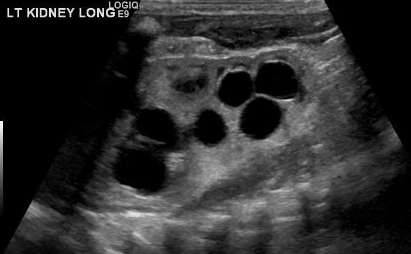
Figura 3 Ultrassonografia renal demonstrando o que pode ser múltiplos cistos renais em um MCDK versus pelvicaliectasia/hidronefrose (HDN).
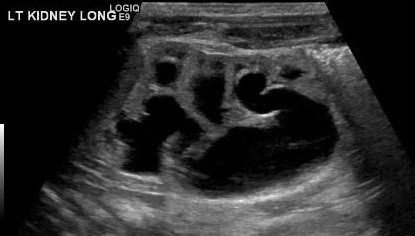
Figura 4 Outra imagem de ultrassonografia renal deste mesmo paciente, demonstrando continuidade entre a pelve e os cálices, compatível com pielocaliectasia/hidronefrose.
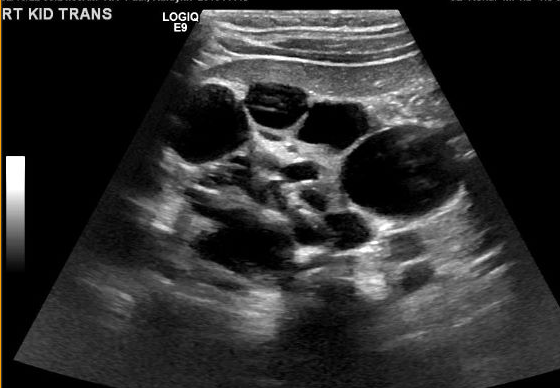
Figura 5 Ultrassonografia renal demonstrando múltiplos cistos não comunicantes de tamanhos diferentes, compatível com MCDK.
Opções de Tratamento, seus Desfechos e Complicações
A história natural do MCDK envolve involução ao longo do tempo, com possíveis complicações incluindo hipertensão e neoplasia maligna.5 No entanto, o risco desta última pode ser baixo.50 Historicamente, os rins acometidos eram removidos cirurgicamente por preocupação com raros casos de degeneração maligna, mas houve uma mudança para o manejo não operatório, com monitorização por ultrassonografia de ambos os rins. As indicações para cirurgia incluem alterações suspeitas de desenvolvimento de tumor de Wilms, efeito de massa, dor, hipertensão e preferência dos pais.5 Pacientes com MCDK e rim contralateral normal devem ser tranquilizados quanto à expectativa de função renal normal em longo prazo, salvo lesão do rim contralateral. Eles também devem ser orientados sobre a importância de medidas de manutenção preventiva, como o uso de equipamentos de proteção ao praticar esportes, e escolhas de estilo de vida saudáveis (dieta e exercício) para mitigar outras causas de doença renal (como hipertensão e diabetes). Para caracterizar o risco de desenvolvimento de doença renal crônica, a vigilância pode adicionalmente incluir exames de imagem do rim contralateral, cistouretrografia miccional para rastreamento de RVU, monitorização da pressão arterial e análise de urina para proteinúria.51
Rim esponjoso medular
O rim esponjoso medular (MSK) é caracterizado por dilatação dos ductos coletores distais, cistos e divertículos, todos restritos às pirâmides medulares.1
Epidemiologia
MSK é encontrada principalmente no início da vida adulta, mas também pode se apresentar em crianças.52 Como muitos indivíduos com MSK são assintomáticos, a incidência não está claramente definida. É mais comum em pessoas com histórico de cálculos renais de cálcio e tem sido associada a síndromes congênitas, como a síndrome de Beckwith-Wiedemann, a síndrome de Ehlers-Danlos, anodontia e doença de Caroli.1
Patogénese
MSK é amplamente considerada não hereditária; no entanto, há casos que parecem ser herdados de forma autossômica dominante, apoiando a hipótese de que a MSK perturba a interface entre o broto ureteral e o blastema metanéfrico.52
Cistos do ducto coletor intrapapilar dilatado e cistos medulares contêm material calcificado ou descamado, conferem ao rim um aspecto esponjoso e são bilaterais em 70% dos casos.1 Os cistos são contíguos aos túbulos coletores e são revestidos por epitélio do ducto coletor.53
Avaliação e Diagnóstico
O diagnóstico pode basear-se em características urográficas de rins aumentados com calcificações ocasionais, especialmente nas papilas, túbulos papilares dilatados que se preenchem com contraste e realce papilar ao contraste com opacificação medular residual.54 A avaliação hepática pode ajudar a distinguir entre MSK e ADPKD, se o diagnóstico estiver incerto.1
Opções de Tratamento, Seus Desfechos e Complicações
O manejo é determinado principalmente pelo tratamento dos cálculos ou das infecções. Adota-se a prevenção de cálculos, incluindo aumento da ingestão de líquidos, dieta com baixo teor de sódio, tiazidas e citrato de potássio.52 As complicações associadas à MSK estão relacionadas à maior incidência de nefrolitíase e à morbidade associada.
Nefronoftise
Nefronoftise (NPHP) é uma causa comum de doença renal crônica em crianças, herdada de forma recessiva, frequentemente associada à doença renal cística medular (MCKD) por apresentarem aspectos radiográficos e histológicos semelhantes; no entanto, a MCKD é herdada de forma autossômica dominante, tem apresentação tardia (vida adulta) e não apresenta envolvimento extrarrenal.5
Epidemiologia
A NPHP é classificada como infantil (1 ano de idade), juvenil (13 anos de idade) ou adolescente (19 anos de idade) com base no defeito genético presente. A NPHP infantil leva à ESRD entre 1–3 anos de idade e está associada a mutações em NPHP2 (também referida como NPHP tipo 2). A NPHP juvenil, a manifestação mais comum, leva à ESRD aos 10–13 anos e está mais frequentemente associada à mutação em NPHP1 (também referida como NPHP tipo 1). Por fim, a NPHP adolescente está associada a mutações em NPHP3 (também referida como NPHP tipo 3).5,55
Patogênese
Embora possa ocorrer esporadicamente, a NPHP é tipicamente herdada de forma autossômica recessiva, sendo as mutações deletérias homozigóticas de NPHP1 no cromossomo 2 a causa mais comum.56 Há muitos genes conhecidos por estarem associados à NPHP (consulte (Tabela 2)5
Tabela 2 Heterogeneidade genética da NPHP.5
| Tipo | Localização cromossômica | Gene mutado |
|---|---|---|
| NPH tipo 1 | 2q13 | NPHP1 |
| NPH tipo 2 | 9q22 | NPHP2 |
| NPH tipo 3 | 3q22 | NPHP3 |
| NPH tipo 4 | 1p36 | NPHP4 |
| NPH tipo 5 | 3q21 | NPHP5 |
| PH tipo 6 | 12q21 | NPHP6 |
| NPH tipo 7 | 16p | NPHP7 |
| NPH tipo 8 | 16q | NPHP8 |
| NPH tipo 9 | 17q11 | NPHP9 |
| NPH tipo 10 | 1q44 | SDCCAG8 |
| PH tipo 11 | 8q22 | MKS3 |
| AHI1 | 6q23 | AHI1 |
Avaliação macroscópica dos rins com NPHP pode variar conforme o tipo, mas eles tendem a ser pequenos, com superfície granular. Histologicamente, exibem fibrose intersticial e infiltração por células mononucleares, alterações tubulares variáveis, incluindo atrofia, espessamento e laminação das membranas basais.57 Os cistos na NPHP originam-se do túbulo contornado distal e dos ductos coletores.57
Avaliação, Diagnóstico e Complicações
Problemas com a capacidade de concentração urinária podem levar a poliúria e polidipsia (desidratação, nictúria) com consequente perda renal de sódio; sintomas de DRC, como fadiga, anorexia, atraso do crescimento; bem como sintomas de osteodistrofia renal. A anemia pode estar presente de forma desproporcional à insuficiência renal. A hipertensão é menos comum, exceto na NPHP tipo 2.5,58 O acometimento extrarrenal inclui fibrose hepática e portal com hepatomegalia, bem como várias outras: síndrome de Senior-Løken (NPHP e degeneração tapetorretiniana), anomalias esqueléticas, deficiência intelectual, ataxia cerebelar, situs inversus, malformação cardíaca ou outras síndromes menos comuns.5
A ultrassonografia é um método comum de identificação, mas pode não detectar todos os casos; a tomografia computadorizada de cortes finos (TC) pode identificá-los.58 Testes genéticos estão disponíveis para o diagnóstico, assim como a histologia renal quando os exames de imagem são inconclusivos. Recomenda-se o rastreamento hepático e ocular para fibrose hepática e síndrome de Senior-Løken.5
Opções de Tratamento e Seus Desfechos
Existem poucos tratamentos eficazes disponíveis para NPHP. O manejo atual inclui medidas de suporte e prevenção para DRC. O transplante em caso de insuficiência renal é preferido, pois a lesão tubular não ocorre no rim do doador.13
Cistos Multiloculares
O cisto multilocular (nefroma cístico) é um crescimento neoplásico benigno que ocorre em adultos e crianças e faz parte de um espectro com o tumor de Wilms.
Epidemiologia
A maioria dos casos ocorre antes dos 4 anos de idade ou após os 30 anos de idade, com predominância masculina na população pediátrica.59
Patogênese
As lesões são volumosas, não comunicantes, bem encapsuladas por tecido fibroso, podem conter tecido embrionário e podem comprimir o parênquima renal normal.5
Avaliação, Diagnóstico e Opções de Tratamento
Cistos multiloculares em crianças mais comumente se apresentam com uma massa abdominal palpável.60 A avaliação é realizada com ultrassonografia ou tomografia computadorizada; contudo, devido à dificuldade, por vezes, de diferenciar entre cistos multiloculares e tumor de Wilms cístico apenas com base em exames de imagem, o diagnóstico definitivo pode ser estabelecido por meio de exame histopatológico após nefrectomia ou nefrectomia parcial.60
Doenças renais císticas adquiridas
A doença renal cística adquirida (ACKD) descreve as alterações císticas bilaterais dos rins no contexto de doença renal em estágio terminal (ESRD) e azotemia e não de doença renal cística hereditária.1
Epidemiologia
Cistos são encontrados em 10% dos pacientes com DRET, aumentando para 44% e 60% aos 3 e 5 anos após o início da diálise.1
Patogênese
Acredita-se que a etiologia seja mediada por toxinas, acumulação de fatores de crescimento, ou por obstrução tubular devida a fibrose, cristais de oxalato, oclusão vascular ou isquemia.1 Os rins são afetados bilateralmente e geralmente são menores do que o normal, enquanto a histologia mostra epitélio plano a cúbico semelhante ao epitélio tubular distal.4
Avaliação, Diagnóstico, Tratamento e Complicações
A maioria dos pacientes com cistos é assintomática, mas pode apresentar dor e/ou hematúria devido a sangramento espontâneo para o interior dos cistos. A ultrassonografia é comumente utilizada para diagnóstico e acompanhamento, enquanto a tomografia computadorizada (TC) ou a ressonância magnética (RM) podem identificar cistos com mais facilidade; no entanto, em pacientes com doença renal em estágio terminal (ESRD) a realização de exames de imagem com contraste intravenoso (IV) deve ser cuidadosamente gerenciada devido aos efeitos nefrotóxicos dos agentes de contraste.1 As principais complicações são hemorragia e transformação neoplásica.4 Recomenda-se ultrassonografia de vigilância, com nefrectomia em um cenário suspeito de neoplasia.5 Assim, o tratamento é conduzido de forma conservadora, com manejo dos sintomas e modulação dos cronogramas de rastreamento ou dos esquemas de diálise.
Síndromes Associadas à Doença Renal Cística
Quistos renais podem ser observados em várias síndromes, sendo as mais comuns a esclerose tuberosa autossómica dominante e Von Hippel-Lindau. Outras, com padrão de herança autossómica recessiva, incluem a síndrome de Meckel, a distrofia torácica asfixiante de Jeune e a síndrome cerebro-hepato-renal de Zellweger.1
Esclerose tuberosa
A esclerose tuberosa é diagnosticada pela presença de dois critérios maiores ou de um critério maior mais dois critérios menores (consulte Tabela 3).61 Múltiplos cistos renais são incluídos como critério menor.61,62 O comprometimento renal é uma causa significativa de morbidade e mortalidade na esclerose tuberosa.63
Tabela 3 Critérios maiores e menores para o complexo de esclerose tuberosa.61
| Critérios maiores | Critérios menores |
|---|---|
| Máculas hipomelanóticas (≥3, com pelo menos 5 mm de diâmetro) | Lesões cutâneas em “confete” |
| Angiofibromas (≥3) ou placa fibrosa cefálica | Fossetas no esmalte dentário (>3) |
| Fibromas ungueais (≥2) | Fibromas intraorais (≥2) |
| Placa de Shagreen | Mancha acrômica retiniana |
| Hamartomas retinianos múltiplos | Múltiplos cistos renais |
| Displasias corticais | Hamartomas não renais |
| Nódulos subependimários | |
| Astrocitoma de células gigantes subependimário | |
| Rabdomioma cardíaco | |
| Linfangioleiomiomatose (LAM) | |
| Angiomiolipomas (≥2) |
Síndrome de Von Hippel-Lindau
A síndrome de von Hippel-Lindau também apresenta muitas manifestações, entre as quais o carcinoma de células renais e os cistos renais são comuns, mas raramente observados em crianças.64
Síndrome de Meckel
Isso pode se apresentar com uma variedade de manifestações, incluindo cistos. A displasia cística está sempre presente.5,65
Conclusões
Cistos renais preenchidos por líquido são encontrados em um grande e heterogêneo grupo de doenças renais císticas. Embora compartilhem uma manifestação comum na forma de cistos renais, essas doenças são diferenciadas por muitos fatores: padrão de herança, idade de início, anomalias renais ou extrarrenais associadas, curso clínico, diferenças histológicas, aspecto em exames de imagem, entre outros. A identificação precisa de um processo patológico pode ser desafiadora quando há sobreposição de sintomatologia. As técnicas de imagem, especialmente a ultrassonografia, há muito tempo têm sido um dos principais meios de identificar muitas dessas doenças. As opções de testes genéticos estão se tornando mais comuns e acessíveis.
A pesquisa contínua leva a avanços em ciência básica e clínica, ajudando a identificar novas doenças, fisiopatologia, modalidades diagnósticas e opções de tratamento.
Pontos-chave
- A distinção entre um cisto simples e um divertículo calicinal pode ser difícil; contudo, é de particular importância, pois um divertículo calicinal é muito mais propenso a causar sintomas e a requerer intervenção. O diagnóstico pode ser facilitado com imagens seccionais com contraste IV e imagens em fase tardia.
- Também é importante distinguir entre MCDK e hidronefrose grave, como no contexto de obstrução da junção ureteropélvica (UPJ). O manejo do MCDK é amplamente observacional, ao passo que há maior probabilidade de que a intervenção seja necessária para hidronefrose ou obstrução de UPJ. O diagnóstico pode ser facilitado com imagem por radionuclídeos.
- O manejo de certas doenças renais císticas, como ADPKD e ARPKD, deve ser realizado em parceria com a nefrologia para facilitar um cuidado médico-renal ótimo.
- A caracterização dos cistos por imagem pré-natal frequentemente ajuda a diferenciar entre possíveis doenças (consulte Figura 6 abaixo).13

Figura 6 Diferenciação de cistos por imagem pré-natal.13
Leituras sugeridas
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
Referências
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- Glassberg KI, Stephens FD, Lebowitz RL. Renal dysgenesis and cystic disease of the kidney: a report of the Committee on Terminology, Nomenclature and Classification, Section on Urology, American Academy of Pediatrics. J Urol Oct 1987; 138 (4 Pt 2): 1085–1092. DOI: 10.1016/s0022-5347(17)43510-5.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364 (16): 1533–1543. DOI: 10.1056/NEJMra1010172.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Cole BR, Conley SB, Stapleton FB. Polycystic kidney disease in the first year of life. J Pediatr Nov 1987; 111 (5): 693–699. DOI: 10.1016/s0022-3476(87)80244-5.
- Gabow PA. Polycystic kidney disease: clues to pathogenesis. Kidney Int Dec 1991; 40 (6): 989–996. DOI: 10.1038/ki.1991.306.
- Proesmans W, Damme B, Casaer P, Marchal G. Autosomal dominant polycystic kidney disease in the neonatal period: association with a cerebral arteriovenous malformation. Pediatrics Dec 1982; 70 (6): 971–975. DOI: 10.1542/peds.70.6.971.
- Ong AC, Wheatley DN. Polycystic kidney disease–the ciliary connection. Lancet Mar 2003; 361 (9359): 774–776. DOI: 10.1016/S0140-6736(03)12662-1.
- Rossetti S, Consugar MB, Chapman AB. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol Jul 2007; 18 (7): 2143–2160. DOI: 10.1681/ASN.2006121387.
- Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol Oct 2004; 15 (10): 2528–2536. DOI: 10.1097/01.ASN.0000141055.57643.E0.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet Apr 1994; 343 (8901): 824–827. DOI: 10.1016/s0140-6736(94)92026-5.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Grantham JJ. Polycystic kidney disease: hereditary and acquired. Adv Intern Med 1993; 38: 409–420.
- Schrier R, McFann K, Johnson A. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol Jul 2002; 13 (7): 1733–1739. DOI: 10.1097/01.asn.0000018407.60002.b9.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med Oct 1990; 323 (16): 1091–1096. DOI: 10.1056/NEJM199010183231602.
- Alzarka B, Morizono H, Bollman JW, Kim D, Guay-Woodford LM. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Front Pediatr 2017; 5 (80). DOI: 10.3389/fped.2017.00080.
- Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers 2018; 4 (1). DOI: 10.1038/s41572-018-0047-y.
- Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatr Nephrol Jan 1989; 3 (1): 43–49. DOI: 10.1007/BF00859625.
- Ward CJ, Hogan MC, Rossetti S. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet Mar 2002; 30 (3): 259–269. DOI: 10.1038/ng833.
- Onuchic LF, Furu L, Nagasawa Y. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet May 2002; 70 (5): 1305–1317. DOI: 10.1086/340448.
- Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol Jun 1997; 11 (3): 302–306. DOI: 10.1007/s004670050281.
- Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics Sep 2014; 134 (3). DOI: 10.1542/peds.2013-3646.
- Blazer S, Zimmer EZ, Blumenfeld Z, Zelikovic I, Bronshtein M. Natural history of fetal simple renal cysts detected in early pregnancy. J Urol 1999; 162 (3 Pt 1): 812–814. DOI: 10.1097/00005392-199909010-00066.
- McHugh K, Stringer DA, Hebert D, Babiak CA. Simple renal cysts in children: diagnosis and follow-up with US. Radiology Feb 1991; 178 (2): 383–385. DOI: 10.1148/radiology.178.2.1987597.
- Bosniak MA. The current radiological approach to renal cysts. Radiology Jan 1986; 158 (1): 1–10. DOI: 10.1148/radiology.158.1.3510019.
- Lee J, Darcy M. Renal cysts and urinomas. Semin Intervent Radiol. Dec 2011; 28 (4): 380–391. DOI: 10.1055/s-0031-1296080.
- Steinhardt GF, Slovis TL, Perlmutter AD. Simple renal cysts in infants. Radiology May 1985; 155 (2): 349–350. DOI: 10.1148/radiology.155.2.3885305.
- Gabow PA, Kimberling WJ, Strain JD, Manco-Johnson ML, Johnson AM. Utility of ultrasonography in the diagnosis of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol Jan 1997; 8 (1): 105–110. DOI: 10.1681/ASN.V81105.
- Peard L, Gargollo P, Grant C. Validation of the modified Bosniak classification system to risk stratify pediatric cystic renal masses: An international, multi-site study from the pediatric urologic oncology working group of the societies for pediatric urology. J Pediatr Urol 2022; 18 (2). DOI: 10.1016/j.jpurol.2021.12.001.
- Bayram MT, Alaygut D, Soylu A, Serdaroğlu E, Cakmakçı H, Kavukçu S. Clinical and radiological course of simple renal cysts in children. Urology Feb 2014; 83 (2): 433–437. DOI: 10.1016/j.urology.2013.08.055.
- Karmazyn B, Tawadros A, Delaney LR. Ultrasound classification of solitary renal cysts in children. J Pediatr Urol Jun 2015; 11 (3). DOI: 10.1016/j.jpurol.2015.03.001.
- O’Kelly F, McAlpine K, Abdeen N, Keays MA, Leonard MP, Guerra LA. The Prevalence, Clinicodemographics, and Outcomes of Incidental and Symptomatic Renal Cysts in a Pediatric Cohort Undergoing Ultrasonography. J Urol 2019; 202 (2): 394–399. DOI: 10.1097/JU.0000000000000264.
- Rediger C, Guerra LA, Keays MA. Renal cyst evolution in childhood: a contemporary observational study. J Pediatr Urol Apr 2019; 15 (2). DOI: 10.1016/j.jpurol.2019.01.006.
- Wulfsohn MA. Pyelocaliceal diverticula. J Urol Jan 1980; 123 (1): 1–8. DOI: 10.1016/s0022-5347(17)55748-1.
- Estrada CR, Datta S, Schneck FX, Bauer SB, Peters CA, Retik AB. Caliceal diverticula in children: natural history and management. J Urol Mar 2009; 181 (3): 1311. DOI: 10.1016/j.juro.2008.10.043.
- Waingankar N, Hayek S, Smith AD, Okeke Z. Calyceal diverticula: a comprehensive review. Rev Urol 2014; 16 (1): 29–43.
- Sahin H, Sarioglu FC, Alaygut D, Akdogan AI, Pekcevik Y. Differentiation of simple renal parenchymal cyst and calyceal diverticulum. Pediatr Int May 2020; 62 (5): 615–623. DOI: 10.1111/ped.14127.
- Casale P, Grady RW, Feng WC, Joyner BD, Mitchell ME. The pediatric caliceal diverticulum: diagnosis and laparoscopic management. J Endourol Sep 2004; 18 (7): 668–671. DOI: 10.1089/end.2004.18.668.
- Long CJ, Weiss DA, Kolon TF, Srinivasan AK, Shukla AR. Pediatric calyceal diverticulum treatment: An experience with endoscopic and laparoscopic approaches. J Pediatr Urol Aug 2015; 11 (4). DOI: 10.1016/j.jpurol.2015.04.013.
- Sripathi V, Mitra A, Padankatti RL, Ganesan T. Robotic treatment of a type 2 calyceal diverticulum in a child: is suture closure and marsupialisation enough for a good outcome? J Robot Surg Dec 2018; 12 (4): 727–730. DOI: 10.1007/s11701-017-0758-1.
- Ding X, Xu ST, Huang YH. Management of symptomatic caliceal diverticular calculi: Minimally invasive percutaneous nephrolithotomy versus flexible ureterorenoscopy. Chronic Dis Transl Med Dec 2016; 2 (4): 250–256. DOI: 10.1016/j.cdtm.2016.11.016.
- Monga M, Smith R, Ferral H, Thomas R. Percutaneous ablation of caliceal diverticulum: long-term followup. J Urol Jan 2000; 163 (1): 28–32. DOI: 10.1016/s0022-5347(05)67965-7.
- Gilbert-Barness E, Potter EL. Respiratory system. 2nd ed., DOI: 10.1001/jama.1997.03540320078045.
- D’Alton M, Romero R, Grannum P, DePalma L, Jeanty P, Hobbins JC. Antenatal diagnosis of renal anomalies with ultrasound. IV Bilateral Multicystic Kidney Disease Am J Obstet Gynecol Mar 1986; 154 (3): 532–537. DOI: 10.1016/0002-9378(86)90597-1.
- Kalyoussef E, Hwang J, Prasad V, Barone J. Segmental multicystic dysplastic kidney in children. Urology Nov 2006; 68 (5). DOI: 10.1016/j.urology.2006.06.024.
- Felson B, Cussen LJ. The hydronephrotic type of unilateral congenital multicystic disease of the kidney. Semin Roentgenol Apr 1975; 10 (2): 113–123. DOI: 10.1016/0037-198x(75)90035-8.
- Welch TR, Wacksman J. The changing approach to multicystic dysplastic kidney in children. J Pediatr Jun 2005; 146 (6): 723–725. DOI: 10.1016/j.jpeds.2005.02.027.
- Sanders RC, Hartman DS. The sonographic distinction between neonatal multicystic kidney and hydronephrosis. Radiology Jun 1984; 151 (3): 621–625. DOI: 10.1148/radiology.151.3.6718720.
- Roach PJ, Paltiel HJ, Perez-Atayde A, Tello RJ, Davis RT, Treves ST. Renal dysplasia in infants: appearance on 99mTc DMSA scintigraphy. Pediatr Radiol 1995; 25 (6): 472–475. DOI: 10.1007/BF02019071.
- Narchi H. Risk of Wilms’ tumour with multicystic kidney disease: a systematic review. Arch Dis Child Feb 2005; 90 (2): 147–149. DOI: 10.1136/adc.2004.051243.
- Mansoor O, Chandar J, Rodriguez MM. Long-term risk of chronic kidney disease in unilateral multicystic dysplastic kidney. Pediatr Nephrol Apr 2011; 26 (4): 597–603. DOI: 10.1007/s00467-010-1746-0.
- Gambaro G, Danza FM, Fabris A. Medullary sponge kidney. Curr Opin Nephrol Hypertens. Jul 2013; 22 (4): 421–426. DOI: 10.1097/MNH.0b013e3283622b86.
- Bernstein J. The classification of renal cysts. Nephron 1973; 11 (2): 91–100. DOI: 10.1159/000180222.
- Gedroyc WM, Saxton HM. More medullary sponge variants. Clin Radiol Jul 1988; 39 (4): 423–425. DOI: 10.1016/s0009-9260(88)80292-7.
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol Jan 2009; 20 (1): 23–35. DOI: 10.1681/ASN.2008050456.
- Hildebrandt F, Otto E, Rensing C. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet Oct 1997; 17 (2): 149–153. DOI: 10.1038/ng1097-149.
- Sherman FE, Studnicki FM, Fetterman G. Renal lesions of familial juvenile nephronophthisis examined by microdissection. Am J Clin Pathol Apr 1971; 55 (4): 391–400. DOI: 10.1093/ajcp/55.4.391.
- Elzouki AY, al-Suhaibani H, Mirza K, al-Sowailem AM. Thin-section computed tomography scans detect medullary cysts in patients believed to have juvenile nephronophthisis. Am J Kidney Dis Feb 1996; 27 (2): 216–219. DOI: 10.1016/s0272-6386(96)90543-0.
- Eble JN, Bonsib SM. Extensively cystic renal neoplasms: cystic nephroma, cystic partially differentiated nephroblastoma, multilocular cystic renal cell carcinoma, and cystic hamartoma of renal pelvis. Semin Diagn Pathol Feb 1998; 15 (1): 2–20.
- Castillo OA, Boyle ET, Kramer SA. Multilocular cysts of kidney. A study of 29 patients and review of literature. Urology Feb 1991; 37 (2): 156–162. DOI: 10.1016/0090-4295(91)80214-r.
- Northrup H, Krueger DA, ITSCC G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol Oct 2013; 49 (4): 243–254. DOI: 10.1016/j.pediatrneurol.2013.08.001.
- Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol Dec 1998; 13 (12): 624–628. DOI: 10.1177/088307389801301206.
- Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc Aug 1991; 66 (8): 792–796. DOI: 10.1016/s0025-6196(12)61196-3.
- Maddock IR, Moran A, Maher ER. A genetic register for von Hippel-Lindau disease. J Med Genet Feb 1996; 33 (2): 120–127. DOI: 10.1136/jmg.33.2.120.
- Salonen R. The Meckel syndrome: clinicopathological findings in 67 patients. Am J Med Genet Aug 1984; 18 (4): 671–689. DOI: 10.1002/ajmg.1320180414.
Ultima atualização: 2025-09-21 13:35