
38: Malformation cloacale
Ce chapitre prendra environ 28 minutes de lecture.
Introduction
Les anomalies anorectales constituent un spectre de malformations congénitales touchant le rectum et l’anus. Les enfants atteints de malformations anorectales (ARM) reçoivent souvent le diagnostic d’« anus imperforé » parce qu’il n’existe pas d’ouverture à l’endroit où l’orifice anal normal devrait se trouver. Cela simplifie à l’excès la pathologie sous-jacente, qui implique fréquemment le système urogénital, la colonne vertébrale et la musculature du plancher pelvien. À une extrémité du spectre se trouvent des anomalies mineures dans lesquelles le canal anal est présent, mais l’anus est antériorisé ou recouvert par la peau périnéale. Dans les malformations plus sévères, le rectum n’atteint pas le périnée et communique plutôt avec l’appareil urogénital.
La manifestation la plus sévère des malformations anorectales chez les filles est une malformation cloaquale dans laquelle le rectum, l’urètre et le vagin se rejoignent pour former un canal commun unique qui s’ouvre sur le périnée. Classiquement, l’orifice périnéal dans une cloaque est antérieur sur le périnée, à l’endroit où s’ouvrirait normalement l’urètre. Les anomalies cloaquales postérieures constituent une variante rare dans laquelle l’orifice est dévié en arrière, le sinus urogénital s’abouchant dans un rectum orthotopique ou juste en avant de celui-ci.1,2
Embryologie et épidémiologie
La malformation cloaquale survient dans environ 1 naissance vivante sur 25 000–50 000. La cause embryologique est imparfaitement comprise, mais implique un échec de la division de la cloaque primitive par le septum urorectal et les plis de Rathke au cours de la gestation précoce.3 Une septation cloaquale anormale est associée à un développement anormal d’autres systèmes d’organes qui se forment dans une proximité temporelle et spatiale étroite, notamment les vertèbres, les uretères, les reins et les dérivés des canaux müllériens. Lorsqu’un enfant naît avec une ARM, il existe un risque de 1 % que les futurs frères et sœurs aient une ARM.4
Dans la malformation cloaquale, le vagin, le rectum et l’urètre sont réunis en un seul canal commun. Un hydrocolpos peut se développer lorsque le vagin se remplit d’urine et de mucus, et la cavité vaginale distendue peut provoquer une obstruction des voies urinaires par compression extrinsèque des uretères ou du col vésical. Les patientes présentant un long canal commun (> 3 cm) sont plus susceptibles d’avoir une dysplasie sacrée et d’autres anomalies congénitales, une musculature sphinctérienne rectale et urinaire déficiente, et sont plus susceptibles de nécessiter une reconstruction chirurgicale plus complexe, y compris une reconstruction vaginale.
Physiopathologie et anomalies associées
Les anomalies urologiques chez les patients présentant des malformations anorectales sont fréquentes et largement sous‑estimées. Elles sont particulièrement prévalentes chez les filles présentant une malformation cloacale. Les pédiatres, les chirurgiens pédiatres et les urologues doivent être conscients de la forte incidence de comorbidités urologiques chez ces patients, et tous les patients devraient bénéficier d’un dépistage des anomalies génito‑urinaires. Les taux rapportés d’anomalies génito‑urinaires associées chez les patients atteints d’ARM varient largement de 18 à 85 %.5 Cette variabilité peut être largement attribuée à des différences dans l’exhaustivité du dépistage. La plupart des séries avec des protocoles de dépistage actifs rapportent une prévalence d’environ 50 % tous types d’ARM confondus, avec des taux d’anomalies urologiques croissants dans les sous‑types d’ARM plus sévères.6 Les malformations cloacales sont le sous‑type le plus sévère de malformation anorectale féminine, et la grande majorité de ces patientes présenteront des comorbidités génito‑urinaires (Tableau 1). Cela souligne l’importance d’inclure les urologues comme membres essentiels de l’équipe multidisciplinaire nécessaire à la prise en charge des patients atteints de malformations anorectales.5,7
Tableau 1 Prévalence des anomalies urologiques chez 712 enfants présentant des malformations anorectales traités au Nationwide Children’s Hospital, Columbus (Ohio). La prévalence de la plupart des anomalies urologiques rapportées augmente à mesure que la sévérité de l’ARM s’accroît. Entre crochets, des plages de prévalence sont rapportées, allant des ARM mineures (périnéales) aux ARM sévères (cloaque à canal long chez la fille, fistule du col vésical chez le garçon). Les anomalies sans différences selon le type d’ARM sont indiquées par un astérisque, et aucune plage n’est rapportée. La dysfonction du bas appareil urinaire et les anomalies müllériennes n’ont pas été incluses dans cette analyse. Adapté de Fuchs et al 2021.6
| Anomalie urologique | Cloaque < 3 cm (n=55) |
Cloaque > 3 cm (n=60) |
|---|---|---|
| >1 diagnostic urologique | 72.7% | 93.9% |
| >2 diagnostics urologiques | 36.4% | 71.7% |
| Hydronéphrose | 47.3% | 76.7% |
| Reflux vésico-urétéral | 21.8% | 31.8% |
| Rein unique | 12.7% | 25.0% |
| Anomalie de fusion rénale | 7.3% | 11.7% |
| Rein duplex | 5.5% | 8.3% |
Anomalies des voies urinaires supérieures
Des anomalies anatomiques concomitantes des voies urinaires supérieures et inférieures surviennent chez 50–60 % des patients présentant des malformations anorectales. Parmi les anomalies fréquemment observées figurent l’hydronéphrose, le reflux vésico-urétéral, la dysplasie rénale, l’agénésie, la duplication et des variantes ectopiques, notamment l’ectopie rénale simple, l’ectopie croisée fusionnée, les reins en fer à cheval et l’ectopie urétérale. Le profil des anomalies des voies urinaires supérieures (ainsi que des anomalies génitales masculines) issu d’une récente étude robuste d’une série unique est résumé dans Tableau 1.6
Outre les anomalies structurelles, il a été rapporté que près de 50 % des filles présentant des anomalies cloaquales développent une insuffisance rénale sévère. Par conséquent, le dépistage précoce de l’atteinte rénale est crucial, tout comme la surveillance et la prise en charge proactives des voies urinaires basses.8
Dysfonction des voies urinaires basses
La dysfonction vésicale est fréquente chez les patients atteints d’ARM. Elle est habituellement associée à une anomalie rachidienne concomitante, toutefois certains enfants présentent une dysfonction vésicale congénitale avec un sacrum osseux et une moelle épinière normaux.9,10 En particulier en cas de malformation cloacale, une dysfonction vésicale acquise peut résulter d’une atteinte neuromusculaire survenue lors de la réparation chirurgicale de la malformation anorectale.11,12 Bien qu’il soit prudent d’adopter une technique chirurgicale méticuleuse et de minimiser l’électrocoagulation monopolaire, certaines atteintes neuromusculaires peuvent être inévitables en raison de la localisation plus médiane du plexus autonome pelvien chez les patients atteints d’ARM.13 Si un dépistage par débitmétrie urinaire est probablement adéquat pour les patients présentant une ARM mineure et une imagerie rachidienne normale, nous préconisons que toutes les filles ayant des anomalies cloacales bénéficient d’un bilan urodynamique.
Anomalies génitales
Les anomalies de l’appareil génital féminin sont moins facilement apparentes à l’examen externe, pourtant les anomalies müllériennes sont très fréquentes chez les filles atteintes d’ARM.14 Les malformations cloacales sont associées à des taux très élevés d’anomalies des organes génitaux internes, avec des chiffres allant de 53 à 67 %.15 Un certain degré de duplication müllérienne est fréquemment observé, allant de cloisons vaginales à une duplication complète de l’utérus et du vagin. Les structures müllériennes peuvent également être hypoplasiques, et l’asymétrie des voies génitales dupliquées est fréquente. Il est essentiel de préciser l’anatomie müllérienne chez les filles atteintes d’ARM. L’hydrocolpos est fréquent et peut entraîner des douleurs, une uropathie obstructive, des infections et une infertilité. Les cliniciens doivent évaluer et traiter l’hydrocolpos chez les nouveau-nés, chez lesquels le liquide vaginal est principalement de l’urine s’accumulant dans les voies génitales, puis de nouveau chez les patientes pubères après la ménarche, chez lesquelles les effluents menstruels peuvent s’accumuler dans le(s) vagin(s). Les taux d’obstruction menstruelle sont rapportés à environ 40 %.8 Chez ces patientes, l’obstruction vaginale peut être congénitale ou due à une sténose vaginale, fréquente après anorectovaginourétroplastie sagittale postérieure (PSARVUP).14,16
Anomalies rachidiennes
Environ un quart à un tiers des patients atteints d’ARM présenteront une pathologie vertébrale ou médullaire associée.17,18,19 Chez les patients présentant spécifiquement une malformation cloacale, de petites séries suggèrent des taux beaucoup plus élevés d’anomalies rachidiennes (plus de 70 %), bien qu’une récente grande étude rétrospective rapporte que 42 % de ces patients présentent une pathologie rachidienne.20,21 Les anomalies fréquentes comprennent l’ancrage médullaire, l’hypoplasie sacrée, l’hémisacrum (qui est associé à une masse présacrée. Les pathologies rachidiennes peuvent entraîner des complications urologiques, neurologiques et orthopédiques.17 L’imagerie rachidienne systématique constitue le standard de soins, et les enfants présentant des pathologies rachidiennes doivent être surveillés pour une vessie neurogène.
Association VACTERL
De multiples syndromes et affections génétiques ont été décrits qui incluent les malformations anorectales parmi leurs caractéristiques. Le plus significatif d’entre eux est l’association VACTERL, bien que de nombreux autres aient été décrits. VACTERL est un acronyme pour les anomalies vertébrales, anorectales, cardiaques, la fistule trachéo-esophagienne, les anomalies rénales et les anomalies des membres. La plupart des cas sont sporadiques plutôt qu’héréditaires. Tous les composants de l’association VACTERL ne sont pas exprimés chez chaque patient. Les plus fréquents sont les anomalies vertébrales, anorectales et rénales. Pour poser le diagnostic du syndrome VACTERL/VACTER, au moins 3 des anomalies doivent être présentes.22 D’autres syndromes qui peuvent être fréquemment associés aux ARMs comprennent MURCS (aplasie des canaux müllériens, aplasie rénale et dysplasie des somites cervicothoraciques), OEIS (omphalocèle, exstrophie, anus imperforé et anomalies rachidiennes).23
Évaluation et diagnostic
Prénatal
La malformation cloacale peut être diagnostiquée en prénatal en raison de l’incidence accrue d’anomalies associées et d’hydrocolpos. L’avantage du diagnostic prénatal est double : les parents peuvent être conseillés, et l’accouchement peut être organisé dans un centre habitué à la prise en charge néonatale des malformations anorectales. Les indices à l’échographie prénatale incluent une dilatation des anses intestinales, un méconium calcifié, l’absence de méconium dans le rectum, un hydrocolpos (généralement identifié comme une structure kystique pelvienne et une vessie mal visualisée), des anomalies rénales, des anomalies du tube neural et l’absence du radius, entre autres. Des anomalies touchant plusieurs systèmes peuvent renforcer la suspicion de VACTERL ou d’un autre syndrome associé à des anomalies anorectales. Certains centres ont eu recours à l’IRM prénatale dans l’espoir de confirmer le diagnostic, bien que celle-ci soit également imparfaite et ne constitue pas le standard actuel de soins.24,25
Examen clinique
Bien que le diagnostic prénatal de malformation cloacale puisse être suspecté à l’imagerie prénatale, la confirmation du diagnostic repose sur l’examen physique en période postnatale. Lors de l’examen périnéal et génital, les urologues doivent évaluer le nombre et la localisation des orifices périnéaux ainsi que le sillon interfessier. Chez les filles, il doit y avoir trois orifices périnéaux distincts (urètre, vagin, anus), avec l’orifice anal centré au sein du complexe sphinctérien anal et séparé du vagin par le corps périnéal. Un orifice périnéal unique chez une fille est pathognomonique d’une malformation cloacale et se situe généralement à l’emplacement attendu de l’urètre. L’aspect du périnée peut varier considérablement, mais la présence d’un orifice unique est l’élément clé à l’examen (Figure 1). Une variante moins fréquente de malformation cloacale, appelée variante cloacale postérieure, présente un orifice périnéal plus postérieur, à l’endroit où se trouveraient le vagin ou l’anus (Figure 2). Parfois, un prépuce clitoridien proéminent donne l’apparence d’une clitoromégalie. En l’absence de corps caverneux palpablement augmentés de volume, cela ne doit pas conduire à un bilan endocrinologique systématique pour des troubles intersexuels.26
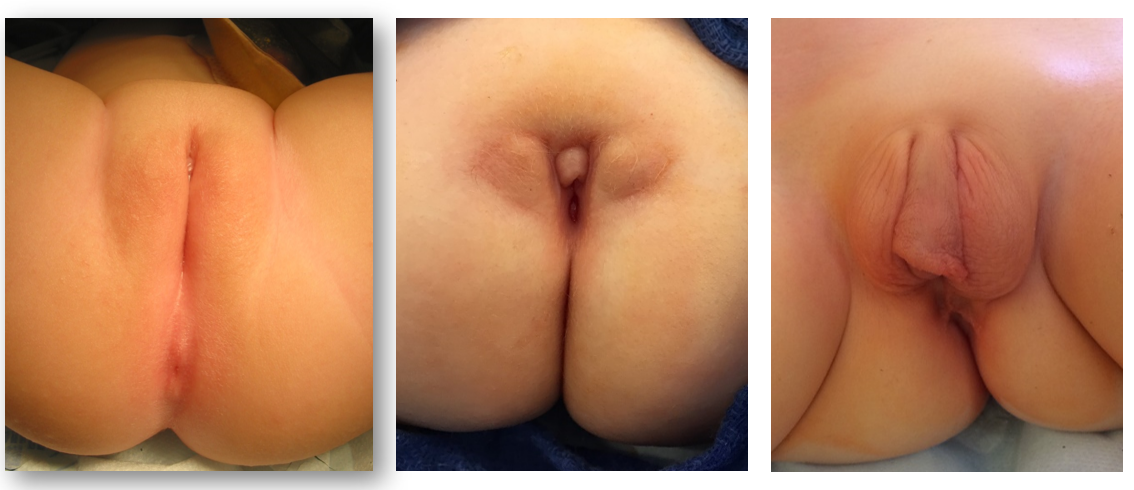
Figure 1 Photographies périnéales de filles présentant une malformation cloacale. L’orifice anal est absent, et l’unique orifice périnéal est situé en arrière du clitoris, près de l’emplacement attendu de l’urètre. Un tissu préputial redondant est fréquent.
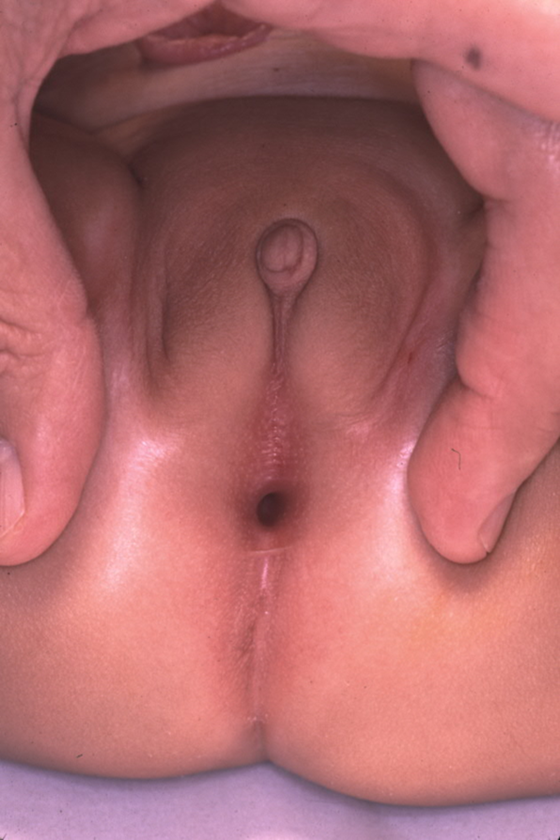
Figure 2 Anomalie cloaquale postérieure, avec l’unique orifice périnéal déplacé en arrière vers l’emplacement attendu de l’anus.
Examens biologiques et imagerie
Le bilan diagnostique ultérieur se concentre sur l’évaluation d’éventuelles anomalies associées.
Échocardiogramme
Ceci est nécessaire pour évaluer les anomalies cardiaques structurelles, en particulier avant l’anesthésie.
Échographie abdominale complète
Rechercher des anomalies rénales, une obstruction des voies urinaires, un hydrocolpos et une duplication müllérienne.
Imagerie rachidienne
Une échographie médullaire doit être réalisée, principalement pour évaluer une moelle attachée. La radiographie standard est utile pour évaluer le sacrum après l’ossification du coccyx. Des radiographies du sacrum en incidences antéro-postérieure et latérale peuvent mettre en évidence des anomalies sacrées telles qu’un hémisacrum et des hémivertèbres sacrées, et permettent d’évaluer le degré d’hypoplasie sacrée. De manière générale, une hypoplasie plus sévère est associée à de moins bons résultats en matière de continence.27 La mesure du “rapport sacré” a été proposée comme indicateur pronostique de la continence fécale, mais ne s’est pas avérée fiable pour prédire la continence urinaire, le développement d’une vessie neurogène ou la nécessité d’un cathétérisme intermittent.28
Pour les patients présentant des anomalies rachidiennes, une évaluation neurochirurgicale et une IRM rachidienne doivent être réalisées ensuite, et ces patients nécessitent un suivi urologique continu pour une vessie neurogène. Les enfants qui se présentent après l’âge de 3–4 mois auront besoin d’une IRM rachidienne, car l’ossification des arcs vertébraux obstrue la fenêtre échographique.
Évaluation de la fonction rénale
Doser la créatinine sérique afin d’évaluer la fonction rénale de base. Noter que l’évaluation de la fonction rénale chez l’enfant est imparfaite, et que l’immaturité rénale chez le nouveau-né peut conduire à une sous-estimation de la fonction rénale, en particulier chez les prématurés. La cystatine C est de plus en plus utilisée en complément de la créatinine sérique.
Des examens d’imagerie complémentaires sont fréquemment réalisés selon les indications cliniques. Dans le contexte d’une hydronéphrose due à un hydrocolpos ou à une distension vésicale, une nouvelle échographie rénale après drainage de l’hydrocolpos doit être réalisée afin de confirmer l’amélioration de l’hydronéphrose. Une scintigraphie rénale ou une uro‑IRM peuvent être envisagées en cas de suspicion de mauvais drainage rénal ou pour déterminer la fonction rénale relative. La cysto‑urétrographie mictionnelle (CUM) peut dépister un reflux vésico‑urétéral, bien que le rôle d’une cysto‑urétrographie mictionnelle préopératoire systématique soit discutable et qu’il puisse être difficile de cathétériser la vessie à l’aveugle. Enfin, bien qu’il existe une forte incidence d’anomalies urodynamiques, les explorations urodynamiques peuvent être différées jusqu’après la reconstruction chirurgicale.
Planification préopératoire
Le reste du bilan est consacré à préciser l’anatomie en vue d’une reconstruction chirurgicale.
Colostogramme
Une colostographie distale à haute pression permettra de déterminer le niveau de la fistule rectale, d’évaluer la longueur de côlon distal disponible pour un abaissement, et de préciser les rapports du rectum avec le sacrum et le coccyx. Après réalisation d’une colostomie, une sonde de Foley est introduite dans la fistule muqueuse et le ballonnet est gonflé délicatement pour prévenir les fuites. Un produit de contraste est ensuite instillé par la sonde afin d’opacifier le côlon distal. La pression du produit de contraste instillé doit être suffisante pour vaincre le tonus des muscles élévateurs de l’anus, et le contraste doit être hydrosoluble et isosmolaire en raison du risque rare de perforation colique. Alternativement, “invertogramme” peut être utilisé pour étudier l’étendue du défaut en cas d’atrésie anale ou rectale. L’anus est marqué à l’aide d’un marqueur radio-opaque, le bébé est placé tête en bas de manière à ce que l’air dans le rectum monte au point le plus élevé, et un cliché radiographique de profil est réalisé. La distance entre l’air et le marqueur radio-opaque indique la distance du rectum à la peau périnéale.
Endoscopie
La cystoscopie et la vaginoscopie sont essentielles à la planification préopératoire pour la réparation d’un cloaque ou d’un sinus urogénital. Celles-ci peuvent être coordonnées avec une colostomie de dérivation ou une autre procédure afin d’éviter une anesthésie inutile. L’endoscopie définira la longueur du canal commun et la distance du col vésical à la confluence, caractérisera le col vésical et le complexe sphinctérien, et évaluera la vessie et les uretères. L’absence d’orifices urétéraux orthotopiques doit faire suspecter un uretère ectopique. La vaginoscopie révélera la présence d’un septum vaginal, d’une duplication vaginale, et la taille du(des) vagin(s) en vue d’une reconstruction ultérieure.
Il est particulièrement important de déterminer la localisation de la confluence vaginale par rapport au col vésical.29 Cette distance correspond à la longueur urétrale et a des implications pour la réparation chirurgicale et les résultats en matière de continence.30 Une longueur urétrale plus courte indique une malformation plus sévère.29
Génitographie
Les génitogrammes sous fluoroscopie sont couramment utilisés pour délimiter les structures urologiques, gynécologiques et rectales. Une sonde de Foley est insérée dans l’orifice périnéal et un produit de contraste est instillé. Les examens fluoroscopiques bidimensionnels conventionnels sont sujets au chevauchement des structures opacifiées et sont donc limités dans leur capacité à définir les rapports tridimensionnels et à obtenir des mesures précises, ce qui souligne la nécessité d’une évaluation endoscopique. De nombreux centres utilisent désormais la tomodensitométrie tridimensionnelle ou l’imagerie par résonance magnétique pour délimiter l’anatomie dans les malformations complexes (Figure 3).31,32
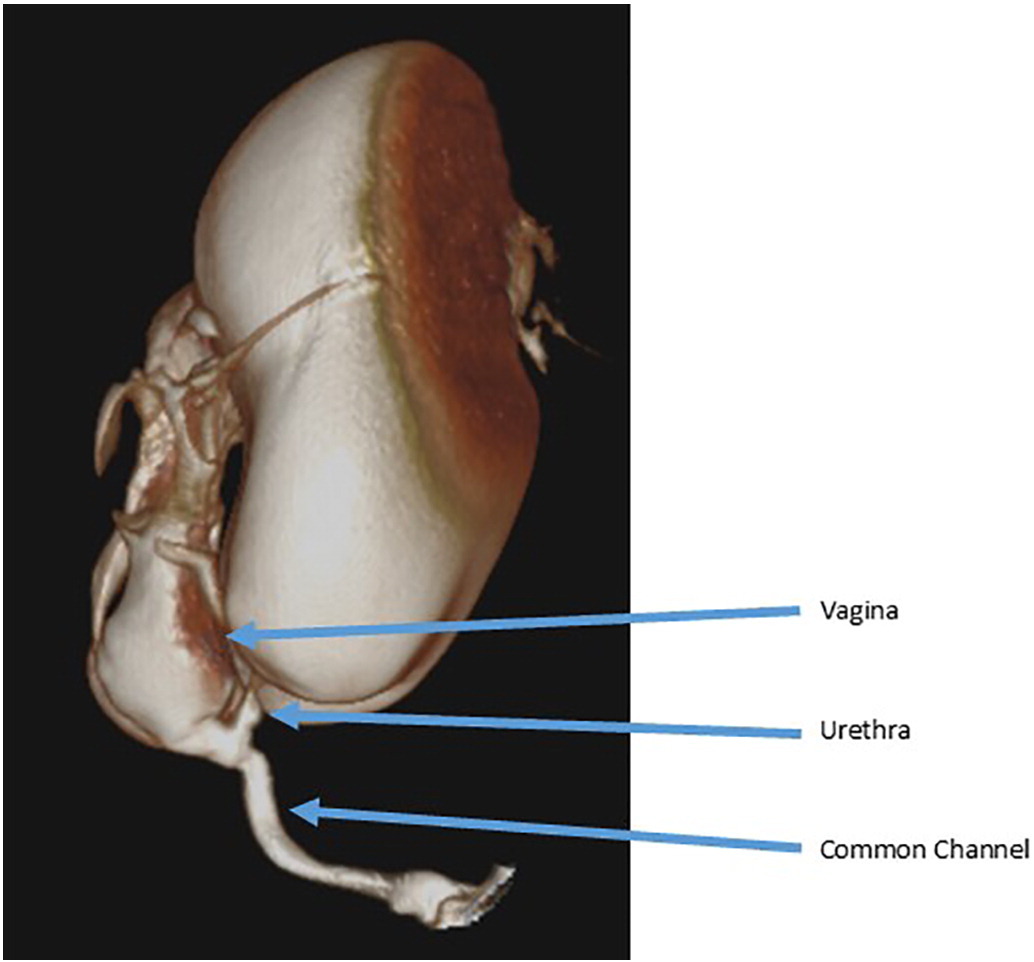
Figure 3 Exemple de cloacogramme tridimensionnel montrant un long canal commun avec une confluence vaginale haute et un urètre court.
Un cloacogramme tridimensionnel (ou une imagerie par résonance magnétique pelvienne standard) est également utile pour délimiter l’anatomie müllérienne et l’ectopie urétérale. Les urologues doivent être conscients d’une affection appelée hémivagin obstrué avec anomalie rénale ipsilatérale (OHVIRA), dans laquelle un hémivagin est obstrué, de sorte que la vaginoscopie mettra en évidence un seul vagin et un seul col utérin. La suspicion d’anomalies müllériennes à l’échographie, discordantes avec l’endoscopie, devrait faire l’objet d’examens complémentaires, et les organes génitaux internes devraient être inspectés directement lorsque l’occasion se présente, par exemple au moment de la colostomie de dérivation initiale.16
Options de traitement, résultats et complications
Période néonatale
Il est essentiel, durant la période néonatale, d’assurer une réanimation adéquate du nouveau-né et la décompression des systèmes d’organes obstrués. La prise en charge initiale doit comporter une colostomie et une fistule muqueuse durant la période néonatale, généralement dans les 24–48 premières heures de vie. Cela permet la décompression du côlon et la séparation du flux fécal des voies urinaires.
Prise en charge de l’hydrocolpos
L’hydrocolpos et une vessie distendue, remplie de liquide, sont fréquents chez les filles présentant une malformation cloacale et se développent lorsque l’urine reflue dans le vagin en raison d’une obstruction urinaire distale. La dilatation vaginale peut être suffisamment sévère pour entraîner une obstruction des voies urinaires supérieures, et l’urine peut refluer par les trompes de Fallope, provoquant une ascite urinaire. Il n’existe pas de consensus quant à la prise en charge de l’hydrocolpos en période néonatale, mais un drainage est nécessaire en cas d’uropathie obstructive. Traditionnellement, le drainage est obtenu par décompression chirurgicale au moyen d’une vaginostomie, d’un cathéter de vaginostomie percutanée ou d’une vésicostomie. Plus récemment, l’option moins invasive du sondage intermittent propre du canal commun a été utilisée.33 Lorsque le canal commun est sondé, la sonde a généralement tendance à entrer préférentiellement dans le vagin plutôt que dans la vessie. Dans une comparaison des deux méthodes de décompression, chirurgicale et CIC, il n’y avait pas de différence dans l’évolution de la créatininémie, ce qui apporte de bonnes preuves que le CIC est une bonne option initiale, non invasive, pour la décompression de l’hydrocolpos.33 En présence d’une cloison vaginale, une vaginostomie peut ne drainer qu’un seul côté du vagin et décomprimer insuffisamment le système. Dans ce cas, une vaginostomie de l’autre côté du vagin ou l’incision de la cloison vaginale peut permettre un meilleur drainage.
Un principe important de toutes les méthodes de décompression de l’hydrocolpos est d’assurer un suivi pour confirmer la réussite du drainage. Après instauration du CIC pour hydrocolpos, l’échographie doit confirmer la décompression du vagin et la résolution de la dilatation des voies urinaires. En cas d’échec, une option plus invasive doit être envisagée. Si l’hydronéphrose persiste malgré la décompression du vagin, un bilan à la recherche d’autres causes d’hydrouretéronéphrose doit être réalisé et une vessie neurogène doit être suspectée.
Gestion vésicale
De nombreuses filles présentant une malformation cloacale n’auront besoin d’aucune intervention pour la vidange vésicale. Si une vessie hostile ou à haute pression avec vidange incomplète est identifiée, une vésicostomie cutanée peut être réalisée afin de permettre une décompression adéquate des voies urinaires. Le sondage intermittent propre est généralement impossible car, comme mentionné plus haut, le cathéter se dirige préférentiellement vers le vagin du fait d’un abouchement urétral fréquemment antérieur. Une vésicostomie peut également être indiquée chez les filles présentant un reflux vésico-urétéral de haut grade ou des infections récidivantes. Là encore, le drainage et la décompression des voies urinaires doivent être confirmés par échographie.
Réparation chirurgicale de la malformation cloaquale
Moment de la réparation
Une fois les systèmes d’organes obstrués décompressés, la réparation chirurgicale primaire de la malformation cloacale n’a pas besoin d’être réalisée immédiatement. Ce délai permet une période d’attachement adéquate entre la famille et l’enfant, mais offre aussi le temps de réunir l’équipe pluridisciplinaire impliquée dans la prise en charge de l’enfant. Cette période permet également d’optimiser la nutrition et la force. Il n’existe pas de consensus sur l’âge optimal de la réparation, mais une réparation < 6 mois après la naissance s’est avérée associée à un taux plus élevé de complications postopératoires, en particulier une désunion de plaie, et la plupart s’accordent à dire que 6–12 mois semble être l’âge idéal pour la réparation primaire.34
Techniques chirurgicales
L’approche chirurgicale est dictée par l’anatomie visualisée au cloacogramme et à la cystovaginoscopie. Deux techniques chirurgicales sont utilisées pour la réparation : la mobilisation urogénitale totale et la séparation urogénitale. Les principes de la réparation chirurgicale consistent à mobiliser l’urètre, le vagin et le rectum jusqu’au périnée afin d’obtenir trois orifices distincts. Cela est réalisé par une incision sagittale postérieure et peut être effectué en association avec une laparotomie si nécessaire. L’abord sagittal postérieur, avec le nourrisson en décubitus ventral, constitue l’abord initial. Ce positionnement permet une meilleure visualisation de l’anatomie par rapport à la position de lithotomie. Dans un premier temps, le rectum est identifié entrant dans le canal commun et mobilisé vers le haut jusqu’au repli péritonéal. À ce stade de l’intervention, une mobilisation urogénitale totale ou une séparation urogénitale est réalisée.
Séparation urogénitale
La séparation urogénitale a été la première technique décrite pour la réparation des malformations cloacales et a constitué l’approche standard popularisée par Hendren (Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10).35 Elle demeure l’approche de choix chez les filles ayant un canal commun > 3 cm de longueur. Dans la séparation urogénitale, le rectum, le vagin et l’urètre sont séparés et amenés individuellement au périnée. Après mobilisation du rectum à partir du canal commun, le vagin est ensuite soigneusement disséqué et libéré de la paroi postérieure de l’urètre, du col vésical et de la vessie. Le défaut de l’urètre au niveau où le vagin a été mobilisé est ensuite fermé en première intention sur une sonde. Cette dissection peut être délicate, et il convient de veiller à ne pas dévasculariser ni dénerver les voies urinaires. Le vagin doit être mobilisé vers le haut autant que possible pour lui permettre d’atteindre la peau périnéale sans tension. Si le vagin n’atteint pas, une voie abdominale peut être nécessaire pour optimiser la mobilisation. Une fois que le vagin atteint la peau, le vagin est abaissé jusqu’à la peau périnéale, fixé en arrière de l’urètre puis latéralement pour créer le néo-introitus. Le corps périnéal est ensuite recréé, et le rectum placé au sein du complexe musculaire. Si, après mobilisation, le vagin n’atteint toujours pas, d’autres techniques de vaginoplastie ont été décrites, telles que la technique de transposition vaginale, une greffe d’interposition intestinale, ou même l’abaissement du vagin natif sous tension, avec l’attente qu’il puisse se rétracter par cicatrisation et nécessiter une introitoplastie ou une vaginoplastie buccale au début de la puberté. La décision concernant la technique de vaginoplastie fait l’objet d’une discussion multidisciplinaire entre la gynécologie, la chirurgie colorectale et la chirurgie urologique, et doit faire l’objet d’une discussion préopératoire avec la famille.
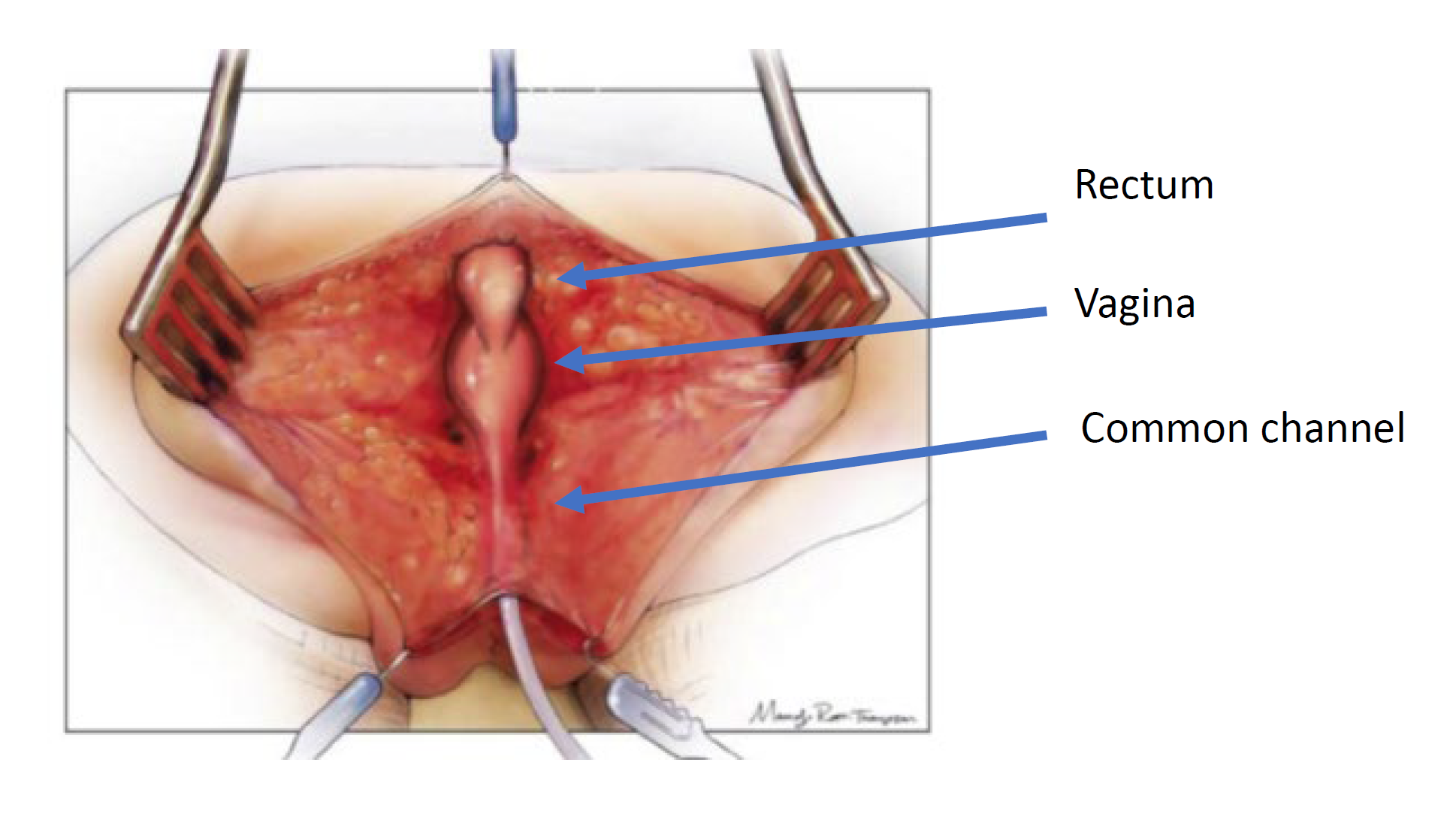
Figure 4 Illustration d’une séparation urogénitale. Le patient est en décubitus ventral, et une incision sagittale postérieure a été réalisée, exposant les faces postérieures du rectum, du vagin et du canal commun. (suite en Figure 5)
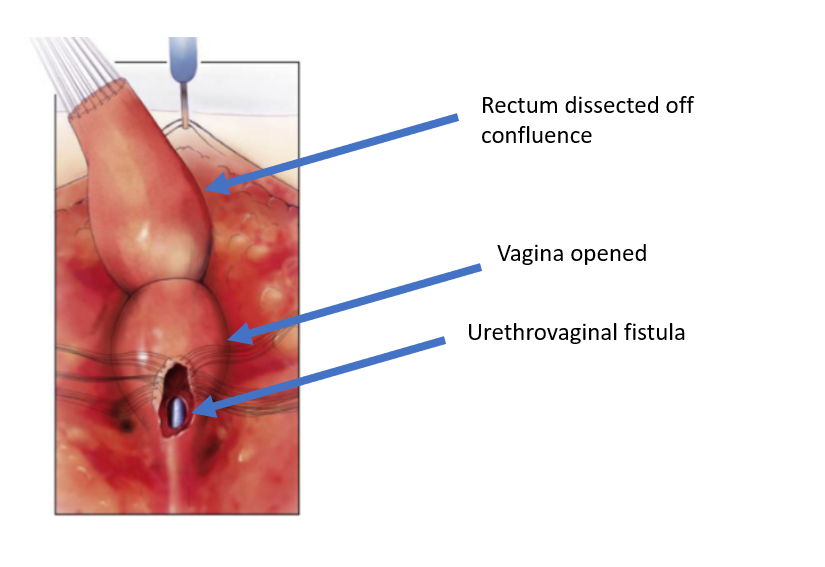
Figure 5 Le rectum a été disséqué du confluent vaginal, laissant une ouverture dans le vagin. (suite de Figure 4, suite dans Figure 6)
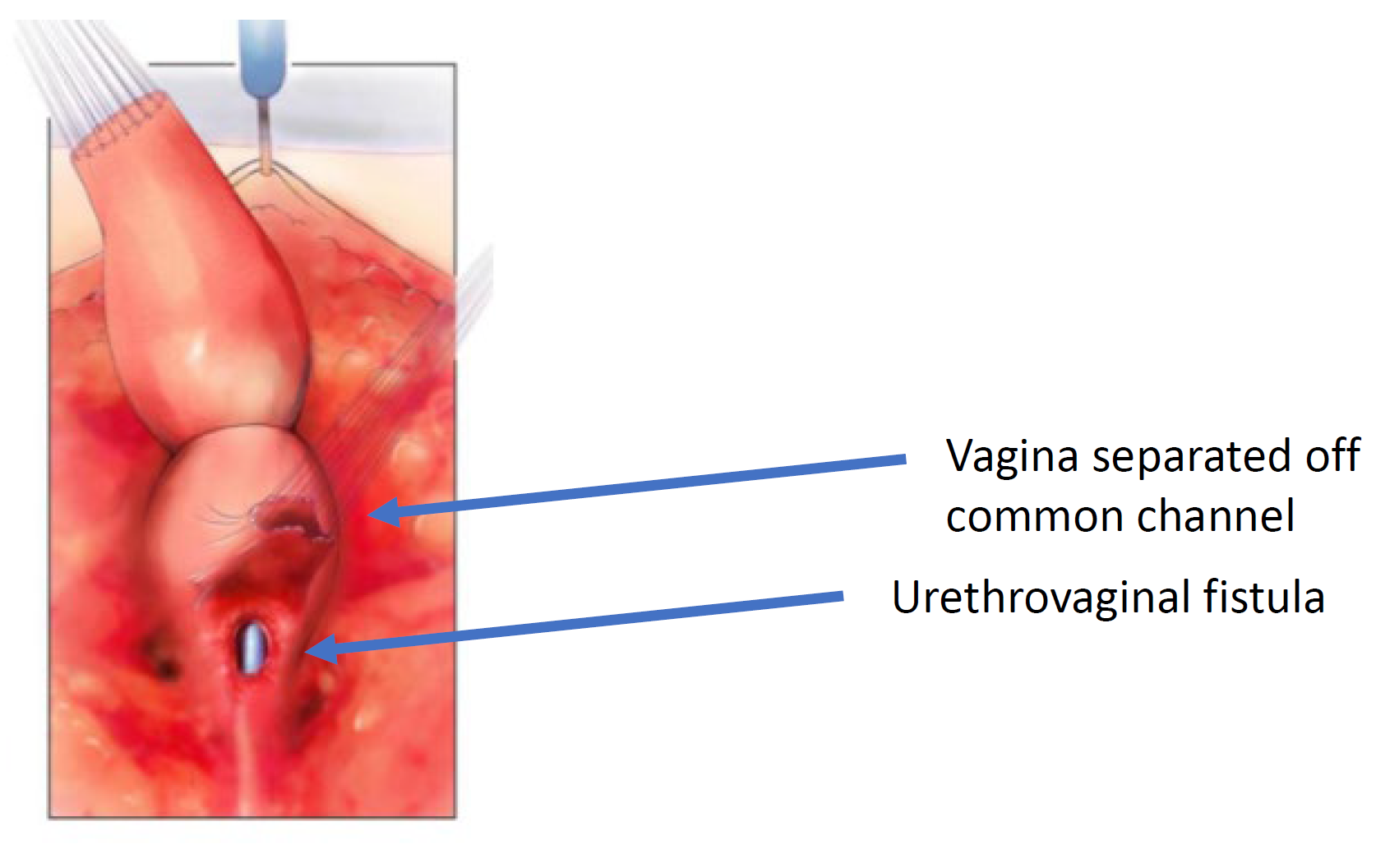
Figure 6 Le vagin a été disséqué du canal commun à sa confluence avec l’urètre. L’orifice à travers lequel le cathéter est visible est la fistule urétrovaginale. (suite de Figure 5, continuation en Figure 7)
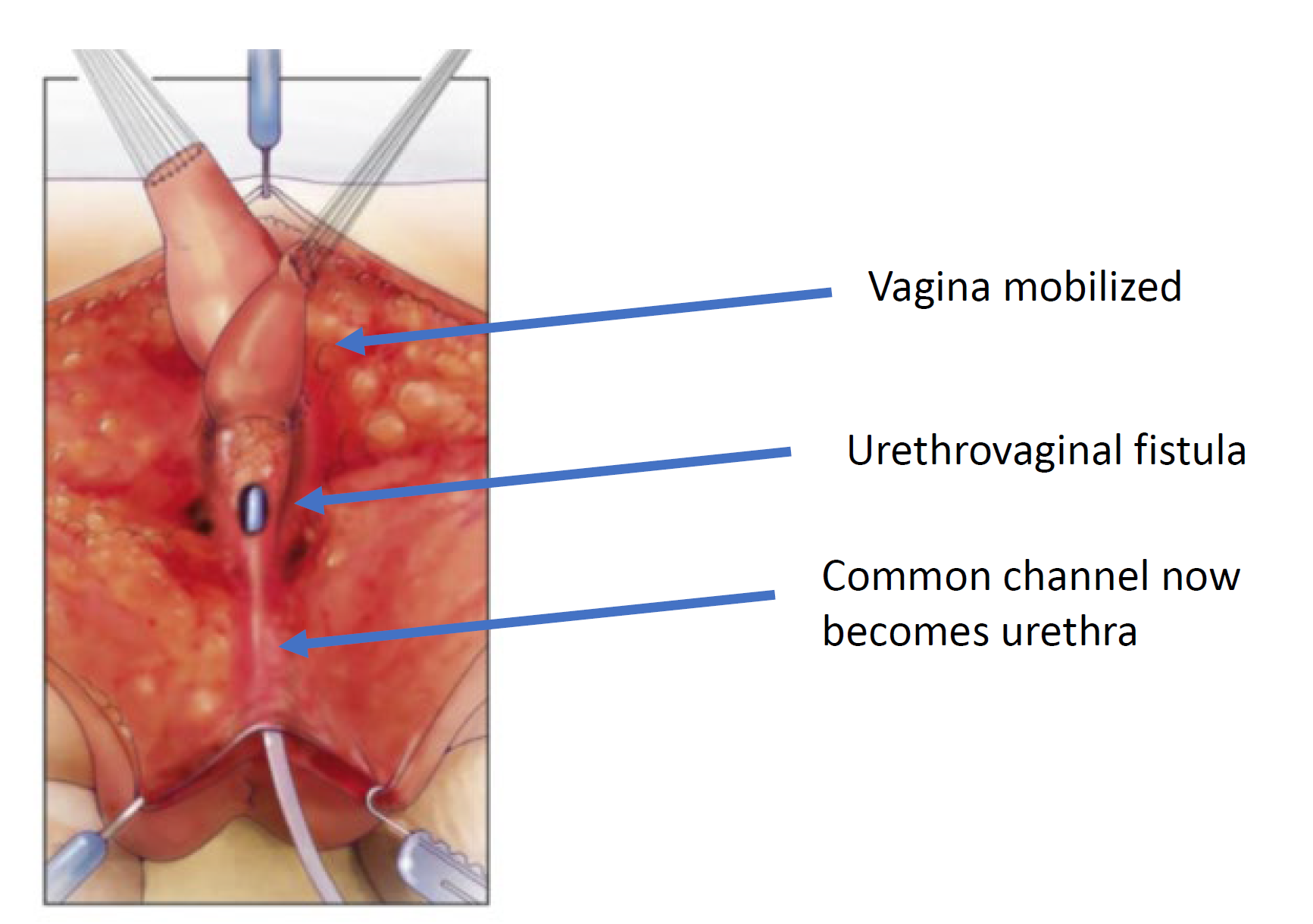
Figure 7 Le vagin est mobilisé proximalement afin d’augmenter sa longueur et son élasticité de sorte qu’il puisse atteindre le périnée. (suite de la Figure 6, suite en Figure 8)
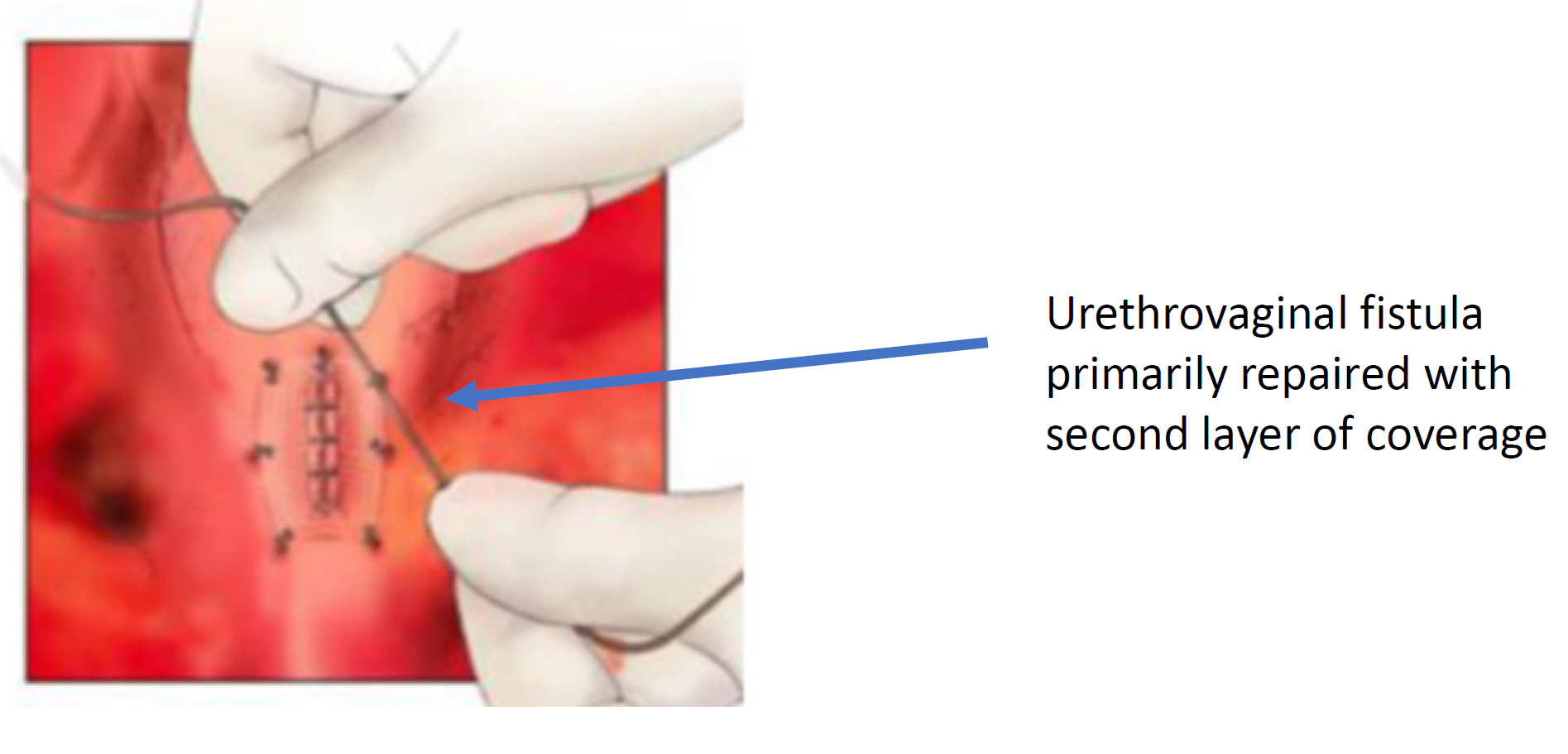
Figure 8 La fistule urétrovaginale est fermée. (suite de la Figure 7)
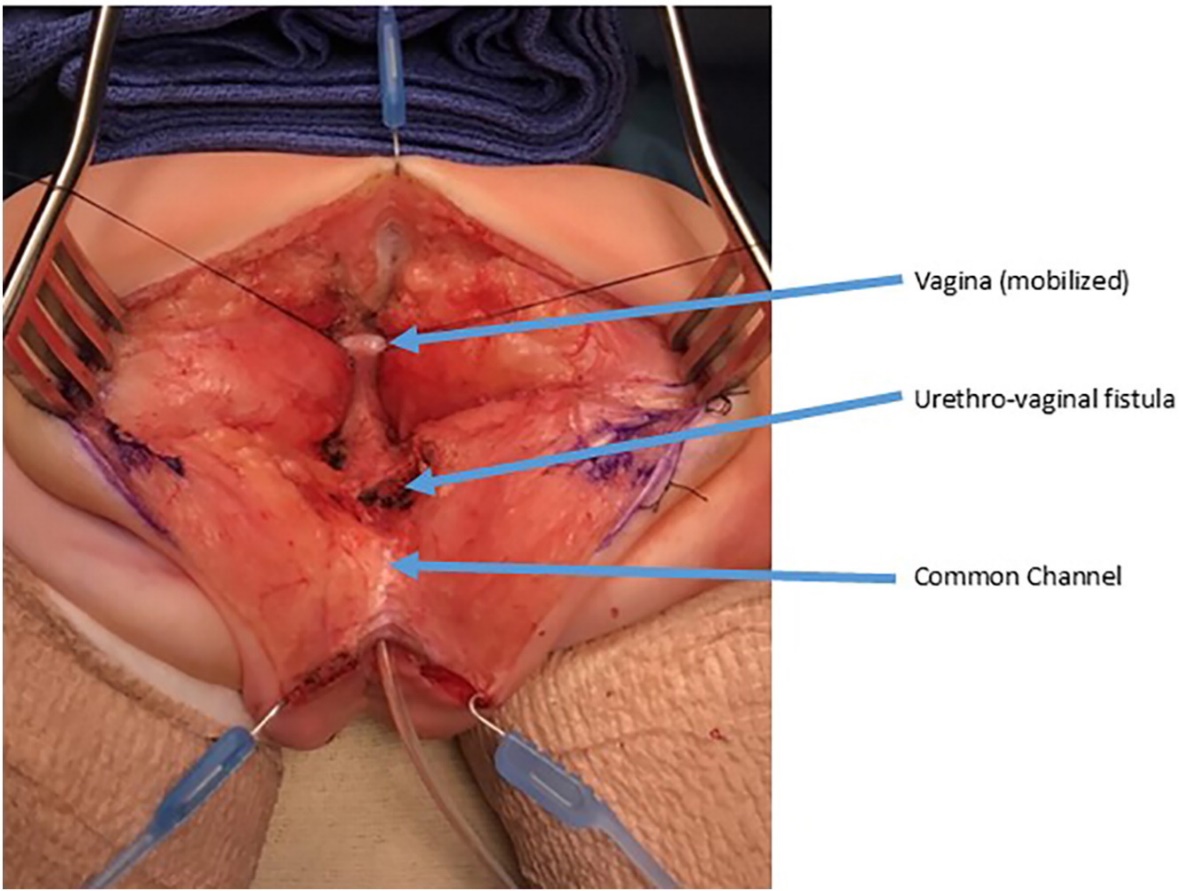
Figure 9 Photographies peropératoires d’une séparation urogénitale. Une incision sagittale postérieure a été réalisée. Une sonde est en place dans le canal commun. Le vagin est maintenu par des fils de traction. La confluence du vagin et de l’urètre est repérée comme une fistule urétrovaginale, qui sera fermée après séparation du vagin de l’urètre. (suite à la Figure 10)
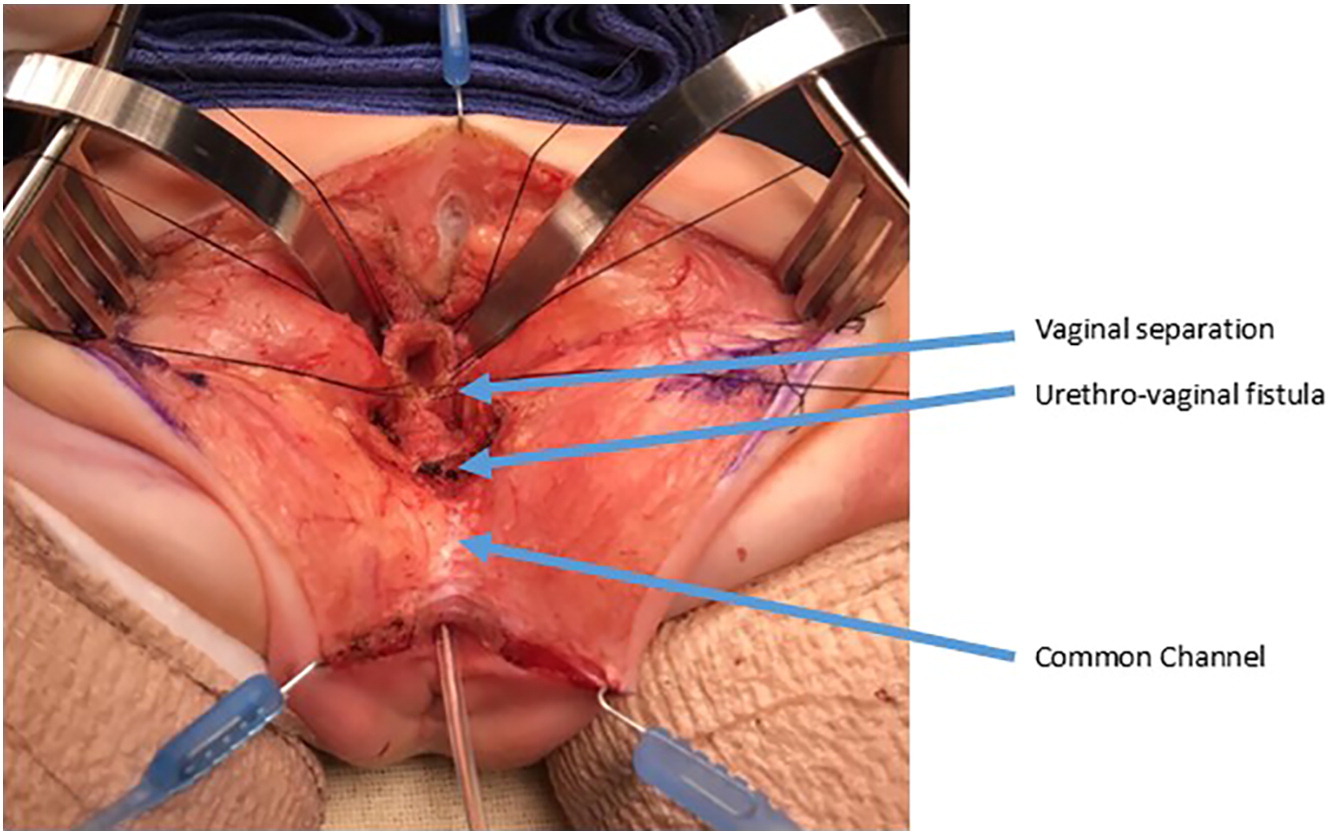
Figure 10 Photographies peropératoires d’une séparation urogénitale. Le vagin a été ouvert et séparé de l’urètre. Le vagin sera ensuite mobilisé proximalement afin de gagner de la longueur pour atteindre le périnée. La fistule urétrovaginale est de nouveau marquée et sera fermée en plans successifs, sur sonde urinaire. (suite de la Figure 9)
Mobilisation urogénitale totale
Une mobilisation urogénitale totale a été introduite pour la première fois pour la réparation cloacale par Peña en 1997 et a été considérée comme une “approche plus simple du cloaque” car elle ne nécessite pas de séparer le vagin des voies urinaires (Figure 11, Figure 12, Figure 13).36 Il s’agit plutôt d’une technique d’abaissement dans laquelle le sinus urogénital est mobilisé jusqu’à ce que la confluence atteigne le périnée, où l’urètre et le vagin sont séparés et anastomosés au périnée. Peña a proposé que cette technique réduirait la survenue de fistules urétro-vaginales et la dévascularisation des voies urinaires. Comme les voies urinaires ne peuvent pas être mobilisées aussi largement que le vagin, il existe des limites à la distance à laquelle le canal commun peut être mobilisé avec cette technique; elle est préférable pour les malformations cloacales avec canal commun court (< 3 cm). Une fois le rectum décollé du canal commun, le canal commun est disséqué circonférentiellement à partir du périnée et ouvert jusqu’à ce que la confluence de l’urètre et du vagin atteigne la peau périnéale sans tension. Le canal commun est fendu et utilisé pour une labioplastie. L’urètre est anastomosé au bord antérieur de l’introitus, juste en arrière du tissu clitoridien. L’urètre et l’introitus vaginal sont anastomosés à la peau sans tension. Le corps périnéal est recréé et le rectum est amené à la peau au sein du complexe musculaire sphinctérien.
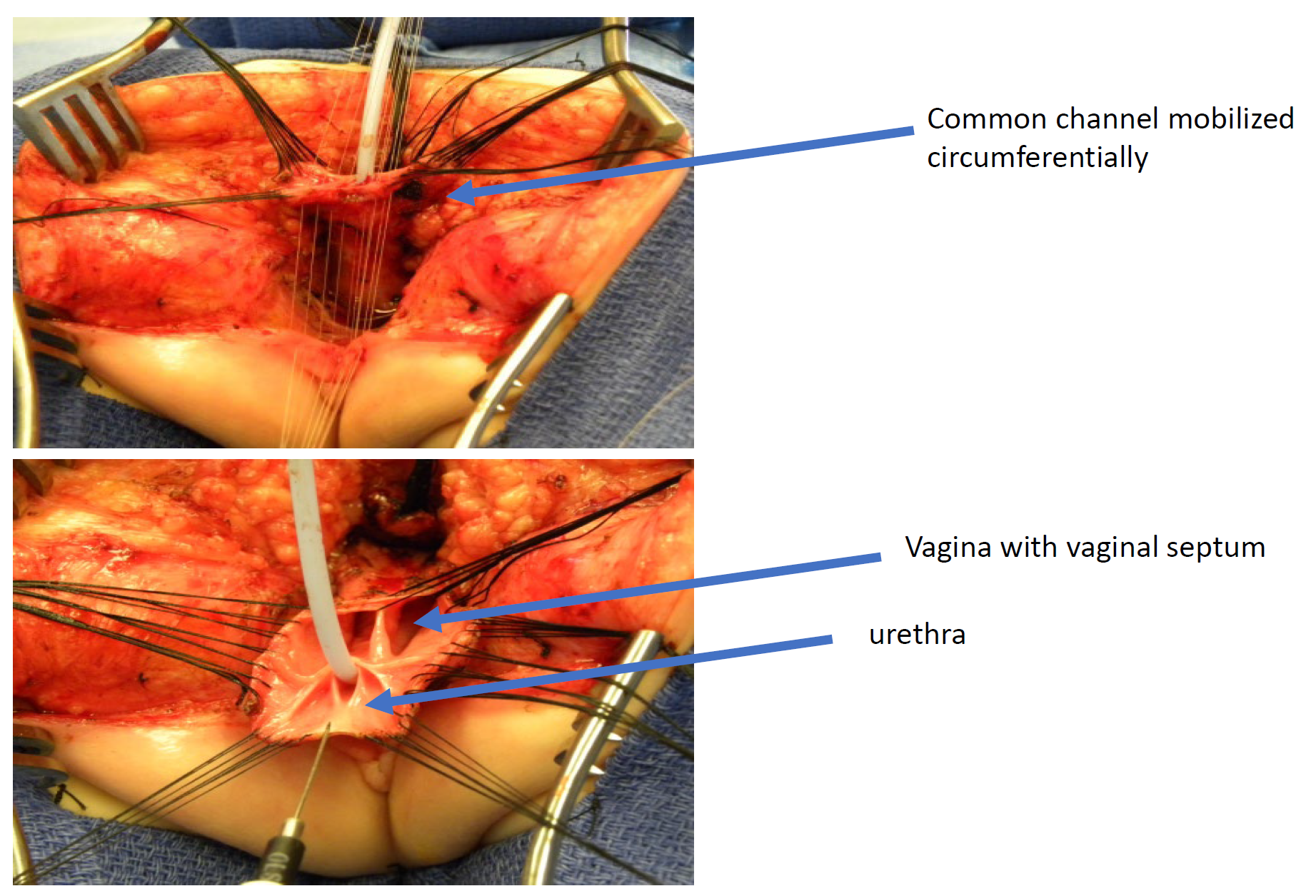
Figure 11 Photographies peropératoires d’une réparation cloacale par mobilisation urogénitale totale (TUM). Une incision sagittale postérieure a été réalisée, et le canal commun et les vagins ont été mobilisés ensemble vers le périnée. Sur cette photographie, le rectum a déjà été séparé.
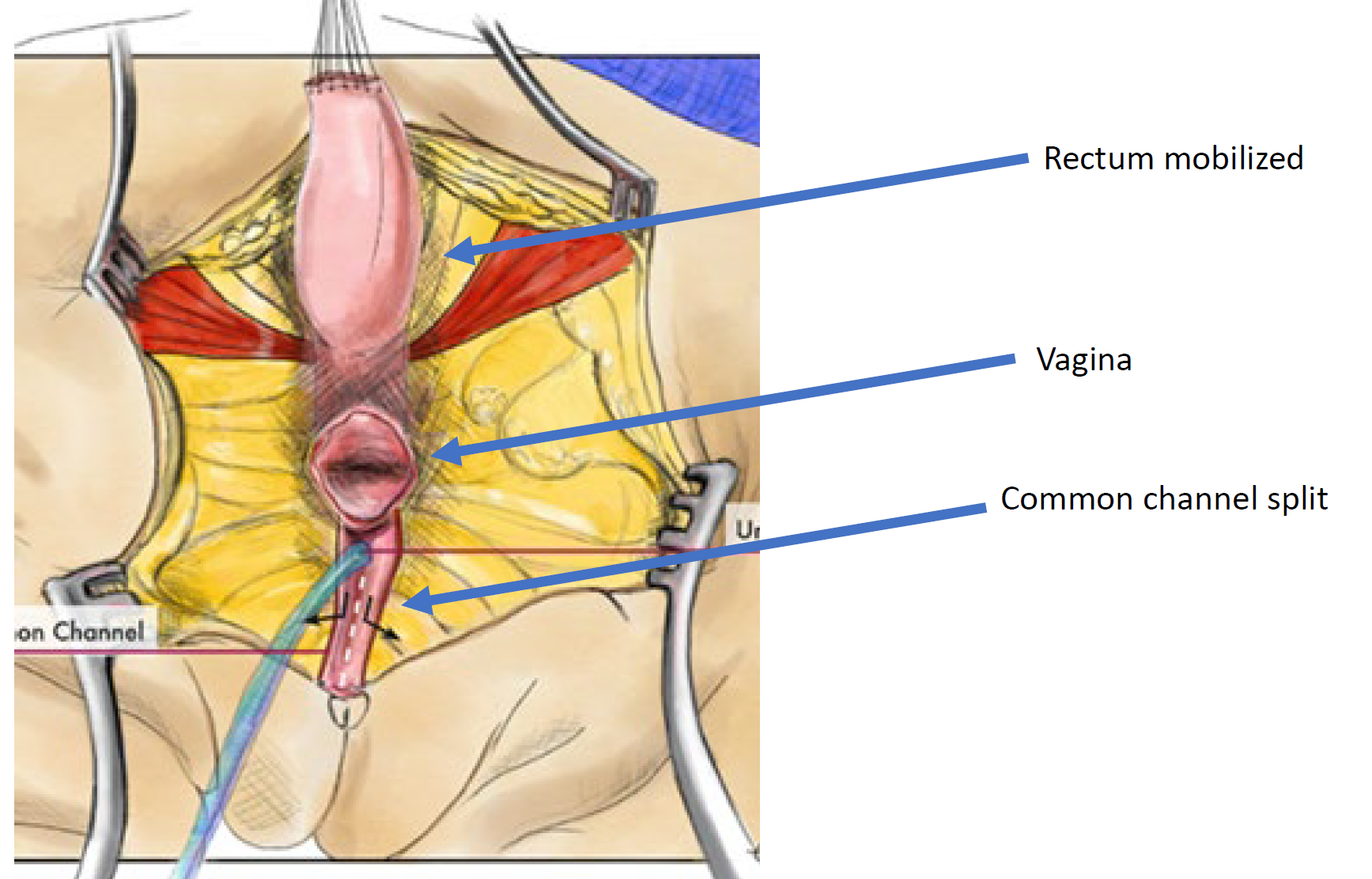
Figure 12 Illustration d’une réparation cloacale par mobilisation urogénitale totale (TUM). La mobilisation est complète une fois que la confluence atteint le périnée et que l’urètre, le vagin et le rectum peuvent être anastomosés séparément au périnée. Le canal commun est scindé, et le tissu redondant est pivoté pour tapisser l’orifice introïtal. (suite à la Figure 13)
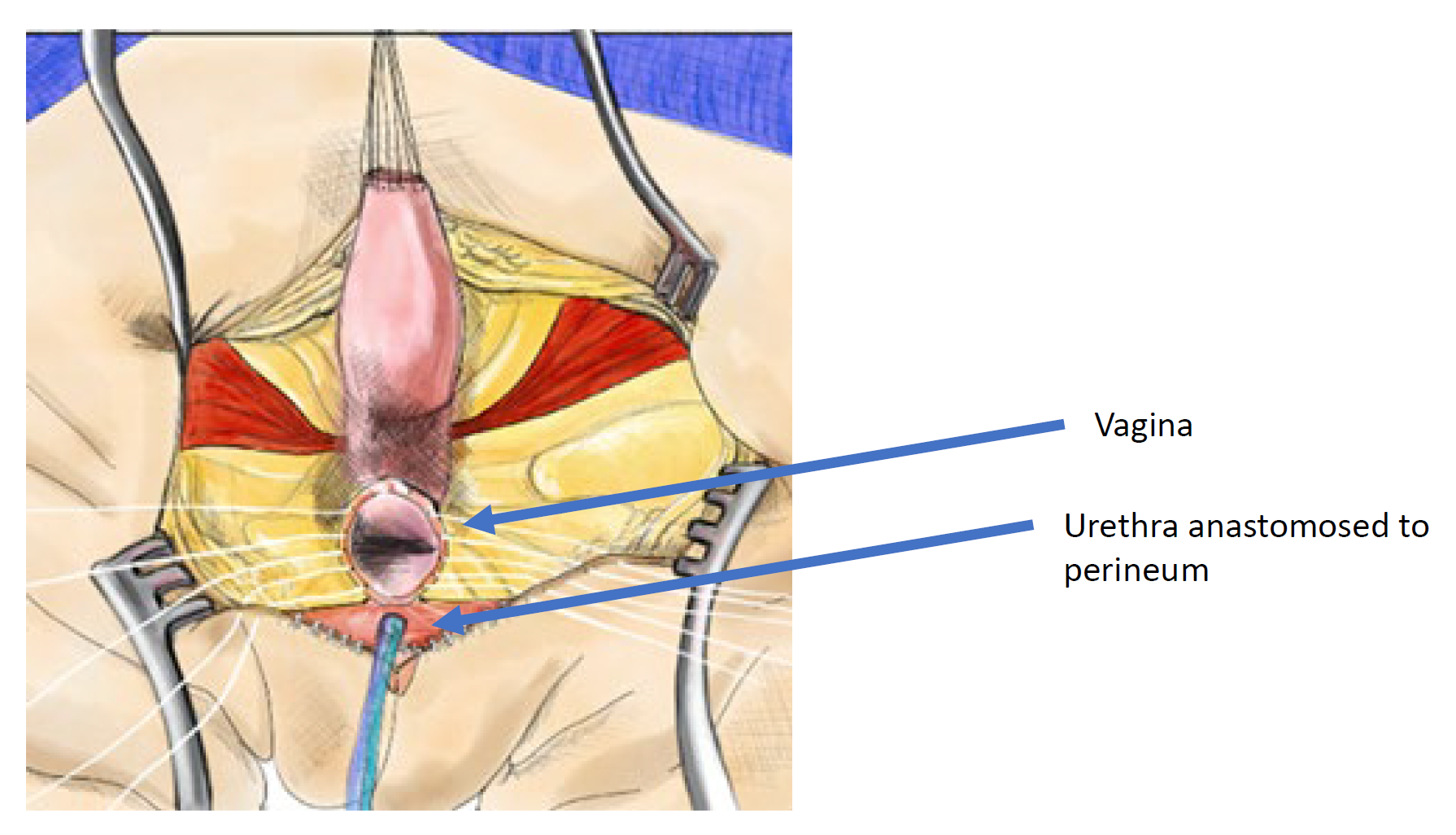
Figure 13 Illustration d’une réparation cloacale par mobilisation urogénitale totale (TUM). La mobilisation est complète une fois que la confluence atteint le périnée et que l’urètre, le vagin et le rectum peuvent être anastomosés séparément au périnée. Le canal commun est divisé, et les tissus redondants sont mis en rotation pour tapisser l’orifice introïtal. (suite de la Figure 12)
Détermination de la technique chirurgicale
Perspective historique
La séparation urogénitale était la technique principale de réparation du cloaque et nécessitait souvent une incision sagittale postérieure associée à une laparotomie. Cette technique est techniquement exigeante et chronophage. Les premiers rapports d’un chirurgien à haut volume d’activité faisaient état de résultats raisonnables, indiquant 58.8% urinant spontanément, 28.4% pratiquant des CIC et 2.8% ayant une dérivation urinaire.35 Cette étude rapporte un “contrôle urinaire” mais aucun détail concernant les accidents urinaires, la vidange ou la fonction rénale n’est rapporté. La description par Peña de la mobilisation urogénitale totale a introduit une approche plus simple de la malformation cloacale. Après un grand nombre de procédures de TUM, il a rapporté des durées opératoires et des hospitalisations plus courtes ainsi qu’une amélioration de la continence urinaire (74%) et de la défécation volontaire.37 Cependant, il a rapporté que chez celles présentant un cloaque avec un long canal commun (> 3 cm), la prise de décision peropératoire était très complexe et que, souvent, après mobilisation complète du canal commun et lorsqu’il n’atteignait pas, le vagin était alors séparé du canal commun mobilisé, laissant l’urètre et le col vésical mobilisés de façon circonférentielle avec une vascularisation compromise et probablement une dénervation. Cela expose l’urètre à un risque de dévascularisation et de nécrose.
Ces deux expériences importantes de la fin des années 1990 ont abouti à un algorithme chirurgical basé sur la mesure du canal commun. Un canal commun court (< 3 cm) serait idéalement traité par une mobilisation urogénitale totale, tandis qu’un canal commun long (> 3 cm) relèverait d’une séparation urogénitale avec ou sans laparotomie ou laparoscopie et une greffe d’interposition intestinale si le vagin n’atteignait pas la peau. Cet algorithme repose fortement sur une mesure précise du canal commun. La mesure peut être réalisée par endoscopie sous vision directe, ou sur un cloacogramme reconstruit en deux dimensions ou trois dimensions (Figure 14). Toutefois, le cloacogramme reconstruit en 3 dimensions s’est révélé la méthode la plus précise pour mesurer le canal commun et permet aux chirurgiens moins expérimentés et aux chirurgiens en formation de comprendre cette anatomie complexe au même niveau que les chirurgiens seniors, améliorant la préparation à l’intervention chirurgicale.38,39
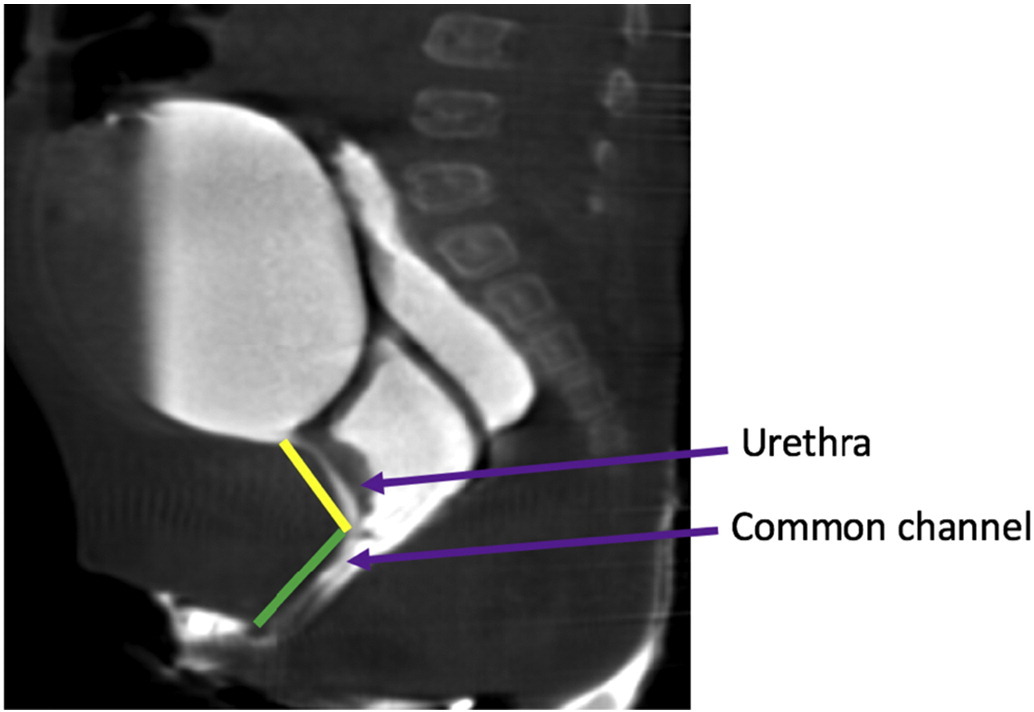
Figure 14 Images IRM T2 sagittales de la malformation cloacale délimitant l’urètre et les longueurs du canal commun.
Développement de l’algorithme chirurgical actuel
Pendant près de 2 décennies, la décision chirurgicale fondée uniquement sur la longueur du canal commun a été le standard de prise en charge. Cependant, à mesure que la prise en charge multidisciplinaire s’est généralisée et que les urologues ont été plus étroitement impliqués aux côtés de la chirurgie pédiatrique dans la prise en charge de ces patients, il a été observé que, si la plupart des filles ayant un canal commun court présentaient un urètre plus long, il existe un sous-type inhabituel de filles avec un canal commun court qui ont un urètre très court. Dans ce sous-groupe de patients, si un TUM était réalisé, il en résulterait un urètre très court, avec un col vésical positionné au niveau du périnée et un très faible potentiel de continence. Suite à cette observation, un algorithme actualisé a été élaboré, intégrant à la fois la longueur du canal commun et celle de l’urètre pour déterminer l’approche chirurgicale Figure 15.13 Dans cet algorithme, si un canal commun est > 3 cm, quelle que soit la longueur de l’urètre, une séparation urogénitale est réalisée. Si un canal commun est < 3 cm et que l’urètre mesure > 1.5 cm, alors un TUM est l’approche opératoire de choix. Dans la variante inhabituelle présentant un canal commun < 3 cm et un urètre < 1.5 cm, ce nouvel algorithme préconiserait la réalisation d’une séparation urogénitale chez l’enfant, maintenant ainsi la position anatomique de la vessie et du col vésical au sein du bassin et évitant de les mobiliser vers le bas et sous le diaphragme pelvien. Cet algorithme chirurgical représentait la première proposition de nouvelle technique chirurgicale en plus de 20 ans depuis la description par Peña du TUM, et il a été largement adopté dans les centres à haut volume. Les résultats de cet algorithme ont été excellents en termes de réduction de la complexité de la prise de décision et des changements de plan peropératoires. Grâce à une évaluation préopératoire précise des mesures de l’urètre et du canal commun, une morbidité urologique significative peut être évitée et on obtient un urètre perméable et cathétérisable chez 97% des patients.40

Figure 15 Algorithme chirurgical proposé pour la malformation cloaquale.13
Prise en charge postopératoire
La prise en charge des plaies et le drainage par sonde sont essentiels après une réparation cloacale par voie sagittale postérieure. Il n’existe pas de consensus quant à la durée du sondage urétral après réparation cloacale, mais elle varie généralement de 1–2 semaines en TUM et jusqu’à 4 semaines lors d’une séparation UG. Certains préconisent un drainage de la vessie par vésicostomie, mais cela n’est pas indispensable. Les incisions sagittale postérieure et introïtale sont à risque de désunion, il est donc crucial d’éviter toute position à califourchon ou le grand écart pendant la cicatrisation. Il est également recommandé d’éviter toute pression périnéale, et que les enfants ne soient pas soutenus par leur périnée ni assis dans des sièges sauteurs ou sur des jouets à califourchon. L’anatomie postopératoire est généralement évaluée par un examen clinique approfondi ou par une cystoscopie et un examen sous anesthésie au bloc opératoire.
Prise en charge vésicale en postopératoire
Après la réparation chirurgicale de la malformation cloacale, il est important de s’assurer d’une vidange vésicale adéquate. La mobilisation chirurgicale et une dysfonction vésicale sous-jacente peuvent entraîner une rétention urinaire ou une vidange incomplète. Cela peut être pris en charge en demandant à tous les enfants d’effectuer un cathétérisme intermittent après le retrait de la sonde et en ajustant le calendrier du cathétérisme en fonction des volumes urinaires. D’autres utilisent une évaluation urodynamique préopératoire pour aider à la prise de décision concernant le CIC. Il n’y a pas de consensus concernant la prise en charge vésicale postopératoire avec cathétérisme. Dans un rapport récent portant sur 18 filles ayant bénéficié d’une réparation cloacale, 44.4% ont nécessité une vidange vésicale assistée (CIC ou vésicostomie) tandis que les autres étaient en couches.41 Les facteurs associés au besoin de CIC après la réparation étaient une grande longueur du canal commun, la séparation UG et la nécessité d’une vidange vésicale assistée avant la réparation chirurgicale. Il est important de noter que la prise en charge par couches en préopératoire ne garantissait pas que la vidange vésicale assistée puisse être évitée en postopératoire.
Prise en charge vaginale en post-opératoire
La perméabilité postopératoire de l’introit vaginal est généralement évaluée au moyen d’un examen approfondi en consultation ou d’un examen sous anesthésie. Si l’introit vaginal devient sténosé tôt dans l’enfance, cela peut être surveillé jusqu’à ce qu’elle soit plus âgée et commence la puberté. Si la sténose est suffisamment sévère pour empêcher l’écoulement des règles, il convient de la prendre en charge avant la ménarche. Une surveillance échographique de l’appareil reproducteur est recommandée à partir de 6–9 mois après le développement du bourgeon mammaire et se poursuit tous les 6–9 mois jusqu’à la ménarche, en évaluant l’épaisseur endométriale dans chaque corps utérin.15 En cas de détection d’un flux menstruel obstrué, une consultation en gynécologie ou en endocrinologie est indiquée afin d’initier une suppression hormonale pour temporiser jusqu’à l’intervention chirurgicale. Les techniques opératoires privilégiées pour les sténoses introïtales ou vaginales comprennent l’introïtoplastie simple ou la vaginoplastie par muqueuse buccale.42 De nombreux centres disposent de spécialistes en gynécologie pédiatrique et de l’adolescente qui assurent ce suivi, bien que des urologues puissent assumer cette prise en charge là où des gynécologues pédiatriques ne sont pas disponibles.
Résultats à long terme
Fonction vésicale
La continence urinaire est difficile à évaluer en raison de la complexité et de l’hétérogénéité de la population. Hendren a rapporté ~60% de mictions spontanées avec un “contrôle urinaire” et Peña a de même rapporté 54% de continence et 46% nécessitant un cathétérisme intermittent propre. Toutefois, aucune de ces études ne fournit de définitions de la continence ni ne rapporte les taux de reconstruction vésicale.35,36 Plus récemment, il y a eu une revue des résultats à long terme portant sur 50 patients de plus de 3 ans ayant une malformation cloacale réparée. Ce groupe rapporte 80% de filles atteignant une “continence sociale”, dont 22% urinent spontanément tandis que les autres réalisent un CIC.43 Ce même groupe comptait 46% nécessitant un nombre moyen de 4.7 interventions chirurgicales pour obtenir la continence. Là encore, la définition de la continence est mal précisée et beaucoup de ceux qui sont “socialement continents” ont eu recours à une chirurgie reconstructrice. Un suivi à long terme est grandement nécessaire dans cette population afin de déterminer les facteurs associés à l’obtention de la continence et des taux de continence fiables.
Fonction rénale
L’insuffisance rénale est une préoccupation constante chez les filles présentant une malformation cloacale. La plus grande série, portant sur 64 patientes, rapporte que plus de 50 % ont un DFG < 80 mL/min/1,73 m2 avec 17 % présentant une insuffisance rénale terminale, 6 % nécessitant une transplantation et 6 % décédées d’une insuffisance rénale.44 Ce même groupe souligne que l’atteinte rénale semble être acquise au fil du temps, puisque 66 % des filles ayant évolué vers une insuffisance rénale chronique avaient un nadir de créatinine sérique normal. D’autres séries plus petites rapportent des résultats nettement plus favorables, avec 66–100 % des filles ayant une fonction rénale normale à 5–9 ans de suivi.45,46,47 Fait intéressant, ces séries sont plus récentes et proviennent toutes d’établissements dotés d’équipes pluridisciplinaires prenant en charge ces patientes. Encore une fois, un suivi à long terme provenant de centres à fort volume évaluant la fonction rénale chez ces patientes est grandement nécessaire.
Points clés
- La malformation cloaquale représente la forme la plus sévère de malformation anorectale féminine
- Les comorbidités urologiques et systémiques sont fréquentes, notamment les anomalies des voies urinaires supérieures, les anomalies müllériennes et la dysfonction vésicale. Une évaluation urologique complète et des soins multidisciplinaires sont primordiaux.
- La prise en charge initiale privilégie un drainage précoce de l’hydrocolpos et une colostomie de dérivation
- La chirurgie de réparation de la malformation cloaquale est appelée anorectovaginorectoplastie sagittale postérieure (PSARVUP). Il existe deux approches chirurgicales principales : la mobilisation urogénitale totale (pour les malformations moins sévères) et la séparation urogénitale (pour les anomalies plus sévères)
- De nombreuses filles nécessitent un cathétérisme vésical. Les données à long terme concernant la continence et la fonction rénale font défaut
Lectures recommandées
- Levitt MA, Peña A. Pitfalls in the management of newborn cloacas. Pediatr Surg Int 2005; 21: 264–269. DOI: 10.1007/s00383-005-1380-2.
- Harris KT, Kong L, Vargas M, Hou V, Pyrzanowski JL, Desanto K, et al.. Considerations and outcomes for adolescents and young adults with cloacal anomalies: a scoping review of urologic, colorectal, gynecologic and psychosocial concerns. Urology 2023. DOI: 10.1016/j.urology.2023.08.047.
Références
- AbouZeid AA, Mohammad SA, Shokry SS, El-Naggar O. Posterior cloaca: A urogenital rather than anorectal anomaly. J Pediatr Urol 2021; 17 (3): 410.e1–410.e7. DOI: 10.1016/j.jpurol.2021.01.014.
- Peña A, Bischoff A, Breech L, Louden E, Levitt MA. Posterior cloaca–further experience and guidelines for the treatment of an unusual anorectal malformation. J Pediatr Surg 2010; 45 (6): 1234–1240. DOI: 10.1016/j.jpedsurg.2010.02.095.
- Thomas DFM. The embryology of persistent cloaca and urogenital sinus malformations. Asian J Androl 2019; 22 (2): 124. DOI: 10.4103/aja.aja_72_19.
- Falcone RA, Levitt MA, Peña A, Bates M. Increased heritability of certain types of anorectal malformations. J Pediatr Surg 2007; 42 (1): 124–128. DOI: 10.1016/j.jpedsurg.2006.09.012.
- Skerritt C, DaJusta DG, Fuchs ME, Pohl H, Gomez-Lobo V, Hewitt G. Long-term urologic and gynecologic follow-up and the importance of collaboration for patients with anorectal malformations. Semin Pediatr Surg 2020; 29 (6): 150987. DOI: 10.1016/j.sempedsurg.2020.150987.
- Fuchs ME, Halleran DR, Bourgeois T, Sebastião Y, Weaver L, Farrell N, et al.. Correlation of anorectal malformation complexity and associated urologic abnormalities. J Pediatr Surg 2021; 56 (11): 1988–1992. DOI: 10.1016/j.jpedsurg.2021.02.051.
- Vilanova-Sanchez A, Halleran DR, Reck-Burneo CA. Re: A Descriptive Model for a Multidisciplinary Unit for Colorectal and Pelvic Malformations. J Urol 2019; 203 (4): 651–651. DOI: 10.1097/ju.0000000000000723.04.
- Warne SA, Wilcox DT, Creighton S, Ransley PG. Long-Term Gynecological Outcome of Patients with Persistent Cloaca. J Urol 2003; 170 (4 Part 2): 1493–1496. DOI: 10.1097/01.ju.0000086702.87930.c2.
- Mosiello GIOVANNI, Capitanucci MARIALUISA, Gatti CLAUDIA, Adorisio OTTAVIO, Lucchetti MARIACHIARA, Silveri MASSIMILIANO, et al.. How to Investigate Neurovesical Dysfunction in Children With Anorectal Malformations. J Urol 2003; 170 (4 Part 2): 1610–1613. DOI: 10.1097/01.ju.0000083883.16836.91.
- Goossens WJH, Blaauw I de, Wijnen MH, Gier RPE de, Kortmann B, Feitz WFJ. Urological anomalies in anorectal malformations in The Netherlands: effects of screening all patients on long-term outcome. Pediatr Surg Int 2011; 27 (10): 1091–1097. DOI: 10.1007/s00383-011-2959-4.
- Jindal B, Grover V, Bhatnagar V. The Assessment of Lower Urinary Tract Function in Children with Anorectal Malformations before and after PSARP. Eur J Pediatr Surg 2009; 19 (01): 34–37. DOI: 10.1055/s-2008-1039027.
- Warne STEPHANIEA, Godley MARGARETL, Wilcox DUNCANT. Surgical Reconstruction Of Cloacal Malformation Can Alter Bladder Function: A Comparative Study With Anorectal Anomalies. J Urol 2004; 172 (6 Part 1): 2377–2381. DOI: 10.1097/01.ju.0000145201.94571.67.
- Davies MRQ. Anatomy of the nerve supply of the rectum, bladder, and internal genitalia in anorectal dysgenesis in the male. J Pediatr Surg 1997; 32 (4): 536–541. DOI: 10.1016/s0022-3468(97)90702-8.
- Cardamone S, Creighton S. A gynaecologic perspective on cloacal malformations. Curr Opin Obstet Gynecol 2015; 27 (5): 345–352. DOI: 10.1097/gco.0000000000000205.
- Breech L. Gynecologic concerns in patients with anorectal malformations. Semin Pediatr Surg 2010; 19 (2): 139–145. DOI: 10.1053/j.sempedsurg.2009.11.019.
- Breech L. Gynecologic concerns in patients with cloacal anomaly. Semin Pediatr Surg 2016; 25 (2): 90–95. DOI: 10.1053/j.sempedsurg.2015.11.006.
- Levitt MA, Patel M, Rodriguez G, Gaylin DS, Pena A. Tethered Spinal Cord in Patients with Anorectal Malformations. Anorectal Malformations in Children 1997; 2 (3): 281–286. DOI: 10.1007/978-3-540-31751-7_18.
- Kim SM, Chang HK, Lee MJ. Re: Spinal Dysraphism With Anorectal Malformation: Lumbosacral Magnetic Resonance Imaging Evaluation of 120 Patients. J Urol 2010; 185 (3): 1094–1094. DOI: 10.1016/s0022-5347(11)60163-8.
- Beaufort CMC de, Groenveld JC, Mackay TM, Slot KM, Beer SA de, Jong JR de, et al.. Spinal cord anomalies in children with anorectal malformations: a retrospective cohort study. Pediatr Surg Int 2023; 39 (1): 281–286. DOI: 10.1007/s00383-023-05440-y.
- Muller CO, Crétolle C, Blanc T, Alova I, Jais J-P, Lortat-Jacob S, et al.. Impact of spinal dysraphism on urinary and faecal prognosis in 25 cases of cloacal malformation. J Pediatr Urol 2014; 10 (6): 1199–1205. DOI: 10.1016/j.jpurol.2014.05.012.
- Garvey EM, Fuller M, Frischer J, Calkins CM, Rentea RM, Ralls M, et al.. Multi-Institutional Review From the Pediatric Colorectal and Pelvic Learning Consortium of Minor Spinal Cord Dysraphism in the Setting of Anorectal Malformations: Diagnosis, Treatment, and Outcomes. J Pediatr Surg 2023; 58 (8): 1582–1587. DOI: 10.1016/j.jpedsurg.2023.04.009.
- Inserm. VACTERL/VATER association. Definitions 2011; 6 (56). DOI: 10.32388/m8pjt5.
- Levitt MA, Peña A. Anorectal malformations. Pediatr Surg Int 2007; 3-3 (2-3). DOI: 10.1007/bf00182758.
- Jacobs SE, Tiusaba L, Al-Shamaileh T, Bokova E, Russell TL, Ho CP, et al.. Fetal and Newborn Management of Cloacal Malformations. Children (Basel) 2022; 9 (6): 888. DOI: 10.3390/children9060888.
- Dannull KA, Browne LP, Meyers MZ. The spectrum of cloacal malformations: how to differentiate each entity prenatally with fetal MRI. Pediatr Radiol 2019; 49 (3): 387–398. DOI: 10.1007/s00247-018-4302-x.
- Bischoff A, Trecartin A, Alaniz V, Hecht S, Wilcox DT, Peña A. A cloacal anomaly is not a disorder of sex development. Pediatr Surg Int 2019; 35 (9): 985–987. DOI: 10.1007/s00383-019-04511-3.
- Vilanova-Sanchez A, Reck CA, Sebastião YV, Fuchs M, Halleran DR, Weaver L, et al.. Can sacral development as a marker for caudal regression help identify associated urologic anomalies in patients with anorectal malformation? J Pediatr Surg 2018; 53 (11): 2178–2182. DOI: 10.1016/j.jpedsurg.2018.03.018.
- VanderBrink BA, Reddy PP. Early urologic considerations in patients with persistent cloaca. Semin Pediatr Surg 2016; 25 (2): 82–89. DOI: 10.1053/j.sempedsurg.2015.11.005.
- Rink RC, Adams MC, Misseri R. A New Classification for Genital Ambiguity and Urogenital Sinus Anomalies. J Urol 2005; 175 (4): 1500–1501. DOI: 10.1016/s0022-5347(05)00847-5.
- Wood RJ, Reck-Burneo CA, Dajusta D. Re: Cloaca Reconstruction: A New Algorithm Which Considers the Role of Urethral Length in Determining Surgical Planning. J Urol 2018; 204 (6): 1368–1368. DOI: 10.1097/ju.0000000000001276.02.
- Patel MN, Racadio JM, Levitt MA, Bischoff A, Racadio JM, Peña A. Complex cloacal malformations: use of rotational fluoroscopy and 3-D reconstruction in diagnosis and surgical planning. Pediatr Radiol 2012; 42 (3): 355–363. DOI: 10.1007/s00247-011-2282-1.
- Halleran DR, Smith CA, Fuller MK, Durhm MM, Dickie B, Avansino JR, et al.. Measure twice and cut once: Comparing endoscopy and 3D cloacagram for the common channel and urethral measurements in patients with cloacal malformations. J Pediatr Surg 2020; 55 (2): 257–260. DOI: 10.1016/j.jpedsurg.2019.10.045.
- Chalmers DJ, Rove KO, Wiedel CA, Tong S, Siparsky GL, Wilcox DT. Clean intermittent catheterization as an initial management strategy provides for adequate preservation of renal function in newborns with persistent cloaca. J Pediatr Urol 2015; 11 (4): 211.e1–211.e4. DOI: 10.1016/j.jpurol.2015.05.014.
- Versteegh HP, Sutcliffe JR, Sloots CEJ, Wijnen RMH, Blaauw I de. Postoperative complications after reconstructive surgery for cloacal malformations: a systematic review. Tech Coloproctol 2015; 19 (4): 201–207. DOI: 10.1007/s10151-015-1265-x.
- Hendren WH. Cloaca, The Most Severe Degree of Imperforate Anus. Ann Surg 1998; 228 (3): 331–346. DOI: 10.1097/00000658-199809000-00006.
- Peña A. Total urogenital mobilization–An easier way to repair cloacas. J Pediatr Surg 1997; 32 (2): 263–268. DOI: 10.1016/s0022-3468(97)90191-3.
- Levitt MA, Peña A. Cloacal malformations: lessons learned from 490 cases. Semin Pediatr Surg 2010; 19 (2): 128–138. DOI: 10.1053/j.sempedsurg.2009.11.012.
- Reck-Burneo CA, Lane V, Bates DG. Re: The Use of Rotational Fluoroscopy and 3-D Reconstruction in the Diagnosis and Surgical Planning for Complex Cloacal Malformations. J Urol 2019; 204 (6): 1367–1368. DOI: 10.1097/ju.0000000000001276.01.
- Gasior AC, Reck C, Lane V, Wood RJ, Patterson J, Strouse R, et al.. Transcending Dimensions: a Comparative Analysis of Cloaca Imaging in Advancing the Surgeon’s Understanding of Complex Anatomy. J Digit Imaging 2019; 32 (5): 761–765. DOI: 10.1007/s10278-018-0139-y.
- Skerritt C, Wood RJ, Jayanthi VR, Levitt MA, Ching CB, DaJusta DG, et al.. Does a standardized operative approach in cloacal reconstruction allow for preservation of a patent urethra? J Pediatr Surg 2021; 56 (12): 2295–2298. DOI: 10.1016/j.jpedsurg.2021.01.011.
- Davis M, Mohan S, Russell T, Feng C, Badillo A, Levitt M, et al.. A prospective cohort study of assisted bladder emptying following primary cloacal repair: The Children’s National experience. J Pediatr Urol 2023; 19 (4): 371.e1–371.e11. DOI: 10.1016/j.jpurol.2023.03.017.
- Leeuwen K van, Baker L, Grimsby G. Autologous buccal mucosa graft for primary and secondary reconstruction of vaginal anomalies. Semin Pediatr Surg 2019; 28 (5): 150843. DOI: 10.1016/j.sempedsurg.2019.150843.
- Warne SA, Wilcox DT, Ransley PG. Long-term Urological Outcome of Patients Presenting with Persistent Cloaca. J Urol 2002; 168(4: 1859–1862. DOI: 10.1097/00005392-200210020-00048.
- Warne SA, Wilcox DT, Ledermann SE, Ransley PG. Renal Outcome in Patients With Cloaca. J Urol 2002; 67 (6): 2548–2551. DOI: 10.1097/00005392-200206000-00055.
- Braga LHP, Lorenzo AJ, Dave S, Del-Valle MH, Khoury AE, Pippi-Salle JL. Long-term renal function and continence status in patients with. Can Urol Assoc J 2007; 1 (4): 371. DOI: 10.5489/cuaj.442.
- DeFoor WR, Bischoff A, Reddy P, VanderBrink B, Minevich E, Schulte M, et al.. Chronic Kidney Disease Stage Progression in Patients Undergoing Repair of Persistent Cloaca. J Urol 2015; 194 (1): 190–194. DOI: 10.1016/j.juro.2015.01.080.
- Rink RC, Herndon CDA, Cain MP, Kaefer M, Dussinger AM, King SJ, et al.. Upper and lower urinary tract outcome after surgical repair of cloacal malformations: a three-decade experience. BJU Int 2005; 96 (1): 131–134. DOI: 10.1111/j.1464-410x.2005.05581.x.
- Levitt MA, Peña A. Pitfalls in the management of newborn cloacas. Pediatr Surg Int 2005; 21: 264–269. DOI: 10.1007/s00383-005-1380-2.
- Harris KT, Kong L, Vargas M, Hou V, Pyrzanowski JL, Desanto K, et al.. Considerations and outcomes for adolescents and young adults with cloacal anomalies: a scoping review of urologic, colorectal, gynecologic and psychosocial concerns. Urology 2023. DOI: 10.1016/j.urology.2023.08.047.
Dernière mise à jour: 2025-09-22 07:59