
22: Exstrophie cloacale
Ce chapitre prendra environ 35 minutes de lecture.
Introduction
L’exstrophie cloacale (CE), communément appelée complexe OEIS (omphalocèle, exstrophie de la vessie, anus imperforé et anomalies rachidiennes), est la manifestation la moins fréquente du complexe exstrophie-épispadias (EEC), toutefois, elle représente la plus grande divergence par rapport à l’anatomie et au développement typiques. En tant que malformation multisystémique, elle nécessite une approche multidisciplinaire dédiée pour obtenir des résultats optimaux pour les patients et leurs familles. Ce chapitre propose une revue de l’exstrophie cloacale dans une perspective moderne, ainsi que des orientations futures pour la compréhension de cette affection lourde.
Embryologie
La théorie prédominante concernant l’EEC est celle d’un défaut d’interposition mésodermique visant à renforcer la membrane cloacale.1 La membrane cloacale est une couche bilaminaire de tissus ectodermique et endodermique située à la partie caudale du disque embryonnaire de la paroi abdominale infra-ombilicale en développement. Dans le développement normal, au cours des 4e et 5e semaines de gestation, l’interposition mésenchymateuse entre ces couches aboutit à la formation des muscles de la paroi abdominale inférieure et du bassin osseux. La poursuite de la descente caudale de ce septum uro-rectal entraîne sa fusion avec la membrane cloacale et, en définitive, la séparation de la cloaque en vessie en avant et rectum en arrière (Figure 1).2 La perforation de la membrane cloacale survient généralement après la fusion avec le septum uro-rectal, vers la 6e semaine de développement, formant des orifices urogénital et anal distincts. Les tubercules génitaux pairs, à l’origine du phallus, migrent ensuite médialement et fusionnent sur la ligne médiane.3
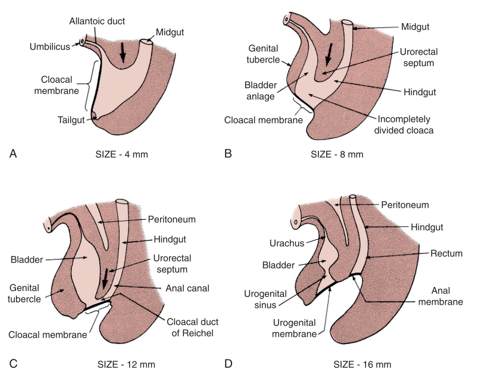
Figure 1 Développement de la membrane cloacale, du sinus urogénital et des orifices anaux. Prolifération mésenchymateuse avec descente caudale du septum urorectal et perforation de la membrane cloacale, aboutissant à la formation des cavités urogénitale et anale.
L’absence de renforcement mésenchymateux et le surdéveloppement de la membrane cloacale qui en résulte exposent à un risque de rupture prématurée de cette membrane. Le moment et le degré auxquels survient la rupture prématurée de cette membrane déterminent le spectre des affections observées dans l’EEC ; la CE est la malformation qui se forme le plus précocement.
Épidémiologie
L’incidence de la CE est d’environ 1 sur 200 000 à 400 000 naissances vivantes4,5 Historiquement, dans la CE, on observe une prédominance homme:femme de 2:1, bien que certaines séries situent ce ratio plus près de 1:1.6,7,8 Avec l’amélioration des modalités d’imagerie et du diagnostic prénatal, toutefois, le nombre de naissances vivantes avec CE a nettement diminué, car les grossesses font l’objet d’une interruption volontaire dans 23 % des cas et jusqu’à 50 % des grossesses s’interrompent spontanément.9
La plupart des cas semblent sporadiques; cependant, des analyses génétiques ont identifié des facteurs causaux possibles. Thauvin-Robinet et al ont décrit une translocation déséquilibrée entre le bras long du chromosome 9 et le chromosome Y, entraînant une délétion 9q34.1-qter comme cause possible, tandis que d’autres ont impliqué des mutations dans un groupe de gènes homeobox, dont HLXB9 et la famille HOX, affectant le développement mésodermique.10
En plus des antécédents familiaux, d’autres facteurs de risque spécifiques à la CE ont été mis en cause. Dans une analyse clinique et moléculaire portant sur 232 familles, un âge parental avancé est apparu comme un facteur de risque possible, avec un âge maternel moyen de 34 ans et un âge paternel moyen de 32 ans (contre 26 et 27 ans, respectivement, selon les données du recensement de 2010). Fait intéressant, 49 % des cas d’exstrophie étaient issus de premières grossesses, ce qui pourrait également renforcer l’effet de l’âge parental avancé.10 Les influences hormonales peuvent également jouer un rôle, Wood et ses collègues ayant identifié une augmentation d’un facteur 10 des naissances d’enfants atteints d’exstrophie chez des mères ayant reçu de fortes doses de progestérone au début du premier trimestre.11 Dans cette même étude, des associations avec les techniques de procréation assistée ont également été rapportées, les enfants conçus par fécondation in vitro (FIV) présentant un risque d’exstrophie multiplié par 7,5.
Actuellement, le facteur environnemental le plus étroitement lié à l’exstrophie cloacale est l’exposition maternelle au tabac, en particulier l’exposition maternelle au tabagisme pendant la période périconceptionnelle et le tabagisme maternel au cours du premier trimestre.12 Pris ensemble, deux points essentiels pour le conseil se dégagent : 1) un conseil approprié concernant l’optimisation des périodes périconceptionnelle et du premier trimestre – durant la période d’organogenèse et 2) la compréhension de l’évolution des points de vue sociétaux concernant la planification familiale (avec des parents accédant pour la première fois à la parentalité à un âge plus avancé) et du besoin potentiel de recourir aux techniques de procréation assistée.
Physiopathologie et anomalies associées
La complexité de la détermination des causes multifactorielles sous-jacentes à la CE se manifeste par l’éventail des anomalies observées dans ce phénotype (Tableau 1). Comme la CE résulte d’événements précoces du développement, elle se manifeste par un plus grand nombre d’anomalies au degré le plus sévère et représente l’extrémité la plus difficile du spectre exstrophie-épispadias. L’expression phénotypique classique de la CE comporte une bande de cæcum extrophié encadrée par deux moitiés vésicales extrophiées, souvent accompagnées d’un segment iléal prolabé, conférant la déformation “trompe d’éléphant” (Figure 2). De plus, l’intestin postérieur est raccourci et se termine souvent en cul-de-sac, entraînant un anus imperforé. Comme les tubercules génitaux ne fusionnent pas sur la ligne médiane, on observe deux moitiés phalliques de part et d’autre d’un diastasis pubien élargi (Figure 3). Le défaut de la paroi abdominale s’accompagne généralement d’une omphalocèle de taille variable.2 En raison de la nature multisystémique de ce défaut, chaque anomalie est décrite plus en détail séparément.
Tableau 1 Exstrophie cloacale et ses anomalies associées.
| Gastro-intestinal | Génito-urinaire | Système nerveux central | Musculo-squelettique |
|---|---|---|---|
| Omphalocèle | Agénésie rénale unilatérale | Moelle épinière attachée | Vertébral (absence ou hémivertèbres) |
| Anus imperforé, atrésie ou sténose anale | Rein pelvien | Myéloméningocèle | Pied bot |
| Syndrome de l’intestin court | Duplication urétérale | Subluxation de hanche | |
| Malrotation intestinale | Hydronéphrose | Pauvreté de la musculature antérieure du plancher pelvien | |
| Duplication intestinale | Cryptorchidie bilatérale | ||
| Hernies inguinales | |||
| Duplication utérine | |||
| Duplication vaginale |
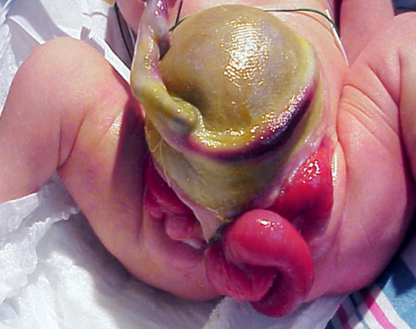
Figure 2 Patient de sexe masculin présentant la forme classique de l’exstrophie cloacale, avec prolapsus d’un segment iléal créant la déformation dite « trompe d’éléphant ».
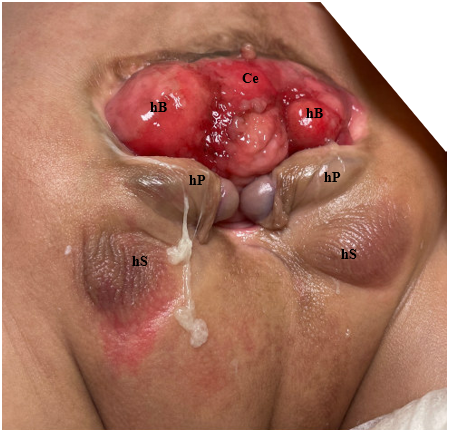
Figure 3 Photographie d’un patient de sexe masculin présentant une exstrophie cloacale. À noter, une colostomie a déjà été réalisée. Ce = caecum; hB = hémivessie; hP = hémiphallus; hS = hémiscrotum.
Appareil musculo-squelettique
Bassin osseux
Les anomalies du bassin osseux et le diastasis pubien associé constituent la caractéristique emblématique de l’exstrophie. En 1995, Sponseller et al ont utilisé la tomodensitométrie (TDM) tridimensionnelle (3D) du bassin osseux chez des patients atteints d’exstrophie et ont mis en évidence des anomalies qui ont modernisé notre compréhension de l’élargissement caractéristique de la symphyse pubienne. Fait notable, ils ont identifié deux grandes catégories d’anomalies, qualifiées d’anomalies rotationnelles et dimensionnelles (Tableau 2).
Tableau 2 Anomalies du bassin osseux.
| Rotationnel | Dimensionnel |
|---|---|
| Rotation externe du bassin postérieur/ailes iliaques | Augmentation du diastasis pubien |
| Rotation externe du segment antérieur du bassin | Raccourcissement du segment pubien antérieur (30%) |
| Rotation coronale de l’articulation sacro-iliaque | Augmentation de la distance entre les cartilages triradiés |
| Rétroversion acétabulaire | |
| Convergence des ailes iliaques | |
| Rétroversion fémorale |
Comparés à des témoins appariés sur l’âge, Sponseller et al ont constaté que, dans l’exstrophie vésicale classique (CBE), la portion postérieure du bassin est en rotation externe en moyenne de 12° de chaque côté, tandis que la portion antérieure du bassin présente une rotation externe en moyenne de 18°. De plus, les branches pubiennes sont plus courtes de 30 %, ce qui, associé à un acétabulum rétroversé, conduit à un diastasis moyen de 4,8 cm en cas de CBE (Figure 4).13 Chez les patients atteints de CE, ces anomalies sont encore plus accentuées, avec une diminution globale de 43 % de la longueur osseuse dans l’ensemble du bassin, avec un diastasis pubien moyen supérieur à 6 cm et une probabilité accrue d’asymétrie entre les côtés droit et gauche du bassin. Pour plus de clarté, un diastasis léger est inférieur à 4 cm, un diastasis modéré est compris entre 4 et 6 cm et un diastasis extrême correspond à tout ce qui est > 6 cm.
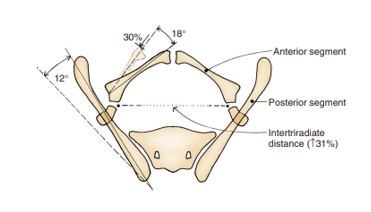
Figure 4 Anomalies osseuses pelviennes observées dans l’exstrophie vésicale classique. Le segment osseux postérieur est en rotation externe de 12˚ de chaque côté, mais la longueur est inchangée. Le segment antérieur est en rotation externe de 18˚ de chaque côté et raccourci de 30 %. La distance entre les cartilages triradiés est augmentée de 31 %. Remarque, les anomalies sont plus sévères dans l’exstrophie cloacale et l’asymétrie est plus fréquente. Utilisé avec l’autorisation du Brady Urological Institute.
Anomalies du plancher pelvien
Les anomalies rotationnelles et dimensionnelles du bassin osseux ont un impact direct sur le développement anormal du plancher pelvien. L’imagerie TDM 3D chez des enfants atteints d’exstrophie a révélé que les muscles élévateurs de l’anus sont positionnés plus en arrière (68 % en arrière/32 % en avant) comparés à des témoins appariés selon l’âge (52 % en arrière/48 % en avant) (Figure 5).14 En plus d’être orienté plus en arrière, l’élévateur de l’anus est également moins concave. Une autre conséquence des déformations du bassin osseux se manifestant au niveau du plancher pelvien est l’aspect plus aplati du muscle puborectal par rapport à sa forme conique habituelle. La pauvreté de la musculature antérieure du plancher pelvien et l’absence de forme conique orientent la technique chirurgicale de reconstruction du bassin osseux et du plancher pelvien, laquelle est abordée séparément.
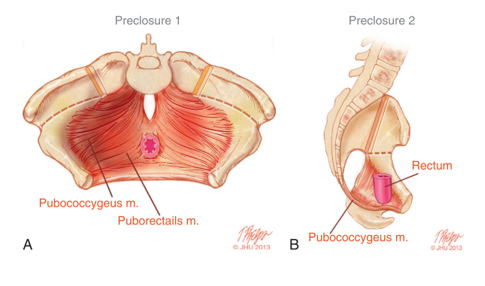
Figure 5 A) Musculature du plancher pelvien avant la fermeture de l’exstrophie, avec les incisions d’ostéotomie marquées. B) Vue latérale de l’anatomie du plancher pelvien montrant le déplacement postérieur des muscles du plancher pelvien derrière le rectum.
Anomalies des membres inférieurs et de la démarche
Des anomalies des membres inférieurs sont constamment observées dans la CE. En raison de l’augmentation du diastasis pubien, de la distance entre les hanches et de la rotation externe des cotyles, les enfants peuvent compenser par une rotation externe des membres inférieurs et une démarche dandinante. Des déformations supplémentaires incluent la torsion tibiale, des déformations en équinovarus et des déformations en calcanéus.15 Malgré ces déformations des membres, l’amplitude articulaire chez ces patients est habituellement préservée et des orthèses correctrices au besoin aident à corriger la démarche avec le temps. À l’âge de 2 ans, la plupart des enfants sont ambulants avec un recours minimal aux dispositifs d’assistance et peu nécessitent un fauteuil roulant durant l’enfance.
Anomalies gastro-intestinales
Les anomalies gastro-intestinales surviennent à un certain degré chez presque tous les patients et augmentent de manière significative la morbidité ainsi que la difficulté de la prise en charge de ces patients, tant en périopératoire en ce qui concerne la nutrition qu’à long terme. L’omphalocèle est caractéristique de cette affection et est retrouvée dans 88–100 % des cas.16,17,18,19 Les omphalocèles peuvent varier considérablement par leur taille et leur contenu, renfermant l’intestin grêle, le foie, ou les deux. La fermeture immédiate du défaut de l’omphalocèle ou la mise en place d’un silo protecteur durant la période néonatale est généralement réalisée afin de prévenir une rupture secondaire tout en assurant un environnement favorable. Dans une série de Davidoff et al incluant 26 patients atteints de CE, tous présentaient une omphalocèle. Parmi ces patients, 19 (73 %) ont bénéficié d’une fermeture primaire, les autres nécessitant une réduction en plusieurs étapes avec un silo prothétique temporaire.16
Le syndrome de l’intestin court, présent dans 25 % des cas, est une cause importante de morbidité nutritionnelle accrue et peut survenir malgré une longueur intestinale normale.4 Une anomalie d’absorption intrinsèque potentielle de l’intestin souligne davantage l’importance d’une nutrition périopératoire optimale chez ces patients et la nécessité de préserver autant que possible la longueur du gros intestin lors de la reconstruction. S’il n’est pas utilisé pour le transit fécal, le reliquat de l’intestin postérieur peut être incorporé lors de la reconstruction de l’appareil urogénital.17 Bien que de nombreuses approches existent pour la prise en charge initiale du tractus intestinal, les travaux de Sawaya et al ont conduit en 2009 à un changement de paradigme avec la tubulisation initiale de la plaque caecale associée à la confection d’une colostomie terminale, car cela a drastiquement réduit la survenue du syndrome de l’intestin court et a permis des procédures de pull-through ultérieures chez les candidats éligibles.20
D’autres anomalies associées du tube digestif sont observées dans 46 % des cas et comprennent des anomalies de duplication, un laparoschisis, une malrotation, une atrésie duodénale et des segments coliques exstrophiés.16,17,21 L’anus imperforé est également fréquemment observé, une série ayant noté sa présence chez 36 des 37 patients.17
Autres anomalies urologiques
Les anomalies des voies urinaires supérieures sont fréquentes, survenant chez 41–66 % des patients.17,18 Il est essentiel de prêter une attention particulière à l’identification et à la compréhension de l’anatomie des voies urinaires supérieures du patient, car une agénésie rénale unilatérale, un rein pelvien ou une hydronéphrose peuvent être observés dans jusqu’à 48 % des cas, avec plusieurs anomalies rénales concomitantes chez 16 % des patients.(missing reference) Les reins en fer à cheval, les anomalies de fusion et les anomalies urétérales, y compris le mégauretère et la duplication, sont rapportés moins fréquemment. À noter que les anomalies urétérales peuvent être très variées, avec une insertion ectopique de l’uretère possible dans le canal déférent chez l’homme et dans l’utérus, le vagin ou les trompes de Fallope chez la femme.19
Anomalies du système nerveux central et de la colonne vertébrale
Le dysraphisme spinal, y compris la moelle attachée, la myéloméningocèle ou la lipomyéloméningocèle, est présent sous une forme ou une autre chez 64 à 100 % des patients, dont 80 % au niveau lombaire.2,18 Les anomalies vertébrales autres que le dysraphisme comprennent des hémivertèbres et une scoliose associée, observée chez 40 % des patients, bien que la scoliose ne progresse pas dans la plupart des cas.15 En raison de cette forte incidence des anomalies spinales et vertébrales, une évaluation par IRM d’un nouveau-né atteint de CE est justifiée, avec intervention neurochirurgicale si nécessaire.
Une atteinte neurologique peut, à terme, affecter la fonction vésicale, la continence urinaire et la motricité des membres inférieurs. Le degré d’atteinte de chacun est tout aussi variable que les anomalies rachidiennes et vertébrales. En outre, la microdissection du bassin d’un nourrisson atteint de CE en contexte post-mortem a mis en évidence une origine et une vascularisation anormales des nerfs autonomes.22 Cette imprévisibilité neurologique complique davantage l’objectif d’aider ces patients à atteindre la continence. Anomalies de la paroi abdominale
La rupture prématurée de la membrane cloacale peut entraîner un large défaut avec absence complète de la paroi abdominale inférieure. Bien que la présentation de ce défaut soit d’emblée évidente, des subtilités nécessitent une attention particulière. Du fait du diastasis pubien élargi et des modifications de l’orientation oblique habituelle du canal inguinal, des hernies inguinales sont observées chez jusqu’à 50 % des patients atteints de CE et devraient être réparées.23
Anomalies affectant les organes reproducteurs et les organes génitaux
Chez le patient de sexe masculin atteint de CE, une séparation complète asymétrique du phallus en deux moitiés, avec une séparation concordante des moitiés scrotales, est typique. Les testicules peuvent être situés dans leurs hémiscrotums respectifs ; toutefois, ils sont souvent non descendus et associés à des hernies inguinales, nécessitant une correction chirurgicale. Chez les patientes de sexe féminin, les corps clitoridiens sont de même largement séparés.
Les anomalies mülleriennes sont également fréquentes dans la CE, avec une duplication utérine observée dans 95 % des cas.19 La duplication partielle prédomine, l’utérus bicorne étant le sous-type le plus courant. Les anomalies vaginales vont de l’agénésie, observée chez 25–50 % des patientes, à la duplication vaginale, qui survient dans jusqu’à 65 % des cas.21 En cas de duplication utérine ou des trompes de Fallope, il convient de privilégier la conservation en vue d’une éventuelle incorporation dans la reconstruction des voies urinaires inférieures.
Anomalies cardiovasculaires et pulmonaires
Rarement, la CE se présente avec des anomalies cardiovasculaires ou pulmonaires mettant immédiatement en jeu le pronostic vital. Des cas rapportés, bien que rares, incluent une pléthore d’anomalies majeures, notamment une duplication de l’aorte et une duplication de la veine cave.23 D’autres ont décrit une tétralogie de Fallot, une atrésie pulmonaire, une sténose de l’artère pulmonaire et une persistance du canal artériel.24
Compte tenu de l’impact associé de cette malformation congénitale sur le développement de presque tous les systèmes d’organes, la prise en charge de ces patients vulnérables devrait être assurée dans un centre ayant l’expérience des techniques chirurgicales et offrant des soins périopératoires et postopératoires multidisciplinaires.
Évaluation et diagnostic
Établir le diagnostic
Compte tenu de l’éventail des malformations associées à la CE, le diagnostic prénatal s’est historiquement avéré difficile, voire insaisissable. Diagnostiquer avec précision la CE et la distinguer de la CBE est primordial pour bien conseiller les familles, d’autant plus que les enfants atteints de CE ont tendance à faire face à davantage de problèmes de santé au cours de leur vie. Le diagnostic prénatal permet une planification susceptible d’optimiser la prise en charge médicale et chirurgicale postnatale. Le premier diagnostic prénatal de CE en 1985 reposait sur trois constatations clés à l’échographie fœtale (fUS) : 1. un grand défaut médian infra-ombilical de la paroi abdominale antérieure, 2. une myéloméningocèle lombo-sacrée, et 3. l’impossibilité de visualiser la vessie.25 Austin et ses collègues ont ensuite affiné cette liste en définissant des critères diagnostiques comme des constatations majeures ou mineures. Observées dans >50% des cas, les constatations majeures comprenaient : la non-visualisation de la vessie (91%), un grand défaut médian infra-ombilical de la paroi antérieure ou une structure kystique de la paroi antérieure (82%), un omphalocèle (77%) et une myéloméningocèle (68%). Les critères mineurs étaient ceux observés dans <50% des cas et comprenaient : des malformations des membres inférieurs (23%), des anomalies rénales (23%), une ascite (41%), un écartement des branches pubiennes (18%), un thorax étroit (9%), une hydrocéphalie (9%) et une artère ombilicale unique (9%).26 Malgré les vastes progrès de l’imagerie en fUS depuis son introduction comme outil diagnostique, on estime qu’à peine 15% des patients ont été diagnostiqués en prénatal par la seule fUS, les constatations pouvant être identifiées de manière incomplète comme un omphalocèle isolé, une CBE ou d’autres défauts de la ligne médiane.
L’avènement de l’imagerie par résonance magnétique fœtale (fMRI) dans le diagnostic prénatal a apporté un complément précieux à l’évaluation de la CE. Comparée à l’échographie fœtale (fUS), l’IRM fœtale (fMRI) offre une visualisation anatomique supérieure lorsque la vessie n’est pas identifiée et peut également aider à évaluer la présence ou l’absence d’un omphalocèle, d’anomalies rachidiennes associées et le sexe, lorsqu’il n’est pas aisément identifié à l’échographie.27,28
Récemment, Weiss et al ont identifié des constatations anatomiques clés en fUS et fMRI pour évaluer leur validité respective dans le diagnostic prénatal de CBE et de CE. Entre 2001 et 2018, ils ont identifié 21 patients ayant bénéficié d’imagerie prénatale. Le diagnostic postnatal était CBE chez 14 patients et CE chez les autres. Quinze des 21 patients disposaient à la fois de fUS et de fMRI pour examen, et l’âge gestationnel médian lors de l’évaluation par imagerie prénatale était de 25 semaines. Parmi les 16 fUS avec interprétations initiales disponibles, le diagnostic prénatal initial était correct dans 12 cas, ce qui correspond à une sensibilité de 69 % du fUS. Les 4 cas de diagnostics prénatals incorrects de CE ont ensuite été reclassés en CBE. Parmi les 18 fMRI incluses dans l’analyse, 16 diagnostics sur 18 étaient concordants (sensibilité de 83 %), et les deux diagnostics prénatals erronés de CE ont été reclassés en CBE. Ces erreurs diagnostiques ont été attribuées à une large plaque vésicale proéminente, avec des anses intestinales en arrière, imitant une omphalocèle contenant de l’intestin. Ils ont conclu que l’identification du point d’insertion du cordon ombilical, tant en fUS qu’en fMRI, était judicieuse pour différencier CBE et CE compte tenu des anomalies de la paroi abdominale.29
Les variantes de l’exstrophie cloacale posent des défis diagnostiques supplémentaires. Bien que extrêmement rare, une variante recouverte de peau est également possible, comme décrit chez 5 des 6 patients dans une petite série de cas.30
Évaluation et prise en charge postnatale immédiate
Après la naissance, il est essentiel de s’assurer que le patient est médicalement stable. Un examen clinique complet est nécessaire pour confirmer les diverses anomalies anatomiques pouvant être présentes (Tableau 1). Mettre en évidence les malformations congénitales associées et leur gravité facilite la prise en charge médicale et chirurgicale. Comme chez les patients atteints de CBE, les segments d’intestin et de vessie exstrophiés sont maintenus humides et protégés par un pansement plastique.31 Une série d’imagerie complète, comprenant des radiographies standards, l’échographie et l’IRM, aide à déterminer la gravité des anomalies associées et facilite l’implication précoce de spécialités consultantes telles que la chirurgie orthopédique pédiatrique, la neurochirurgie pédiatrique et la chirurgie pédiatrique générale. Compléter l’équipe pluridisciplinaire par l’avis et le soutien du service social et des services de nutrition est essentiel pour la prise en charge du nourrisson atteint de CE et de sa famille.
Assignation du genre
Une fois que le nourrisson est médicalement stable et que l’étendue complète de la pathologie est comprise, une discussion honnête et réfléchie concernant l’attribution du sexe est nécessaire. Dans de nombreux cas, cela peut nécessiter une consultation en endocrinologie pédiatrique, en psychologie de l’enfant ou en psychiatrie pédiatrique. Toute décision relative à l’attribution du sexe ne devrait être prise qu’après réalisation d’un caryotype, une discussion approfondie et un accompagnement parental approprié.
Compte tenu de l’expression phénotypique typique, avec une large séparation des corps péniens et du scrotum et une taille réduite des corps caverneux chez les garçons atteints de CE, les premiers rapports prônaient une réassignation de genre universelle des garçons 46, XY en femmes fonctionnelles.32 Cela a orienté les premiers principes de la prise en charge, incluant une orchidectomie bilatérale ainsi qu’une reconstruction phallique en clitoris fonctionnel et une vaginoplastie, soit précoces, soit différées.
Les effets à long terme de cette pratique font actuellement l’objet d’un débat intense. Des patients vivant plus longtemps avec cette affection offrent un aperçu des implications psychosociales de cette pratique et du rôle potentiel du génotype et du milieu hormonal intra-utérin. Dans une cohorte de 29 sujets de sexe masculin atteints de CE ayant subi une réassignation vers le genre féminin, les 29 patients ont montré un basculement prédominant vers le masculin dans le développement psychosexuel, malgré l’absence de toute poussée hormonale pubertaire.33 D’autres séries, toutefois, n’ont montré aucune différence de comportement ou de problèmes psychosociaux, bien qu’un cas de masculinisation ait été rapporté chez un patient 46XY converti de genre en raison d’un testicule ectopique.34,35 Les attitudes actuelles privilégient, si possible, l’assignation d’un genre concordant avec le caryotype. Cela a été étayé par une enquête auprès d’urologues pédiatriques, dans laquelle les deux tiers privilégiaient une reconstruction concordante avec le genre.36
Prise en charge chirurgicale et résultats
Prise en charge initiale
La fermeture précoce de l’omphalocèle au cours de la période néonatale est recommandée pour
prévenir une rupture prématurée; toutefois, cette mesure n’est réalisée qu’après la prise en charge des impératifs neurochirurgicaux. Au moment de la fermeture initiale de l’omphalocèle, l’une des 3 approches est généralement employée pour la dérivation intestinale et la prise en charge de l’intestin postérieur, notamment la création d’une iléostomie avec résection de l’intestin postérieur, la mise en place d’une iléostomie avec fistule muqueuse de l’intestin postérieur, ou la tubulisation du cæcum avec création d’une colostomie terminale.
Historiquement, la dérivation intestinale initiale reposait sur une iléostomie avec résection de l’intestin postérieur, cependant cela entraînait plusieurs conséquences non intentionnelles. Premièrement, cela induisait universellement un syndrome de grêle court et rendait moins probable une reconstruction ultérieure du tractus gastro-intestinal. Deuxièmement, les iléostomies à haut débit prédisposaient les enfants à une augmentation des hospitalisations avec des déshydratations récurrentes et des troubles hydroélectrolytiques.18,20 L’acidose induite secondaire à un débit iléostomique élevé altère l’homéostasie calcique et module directement l’axe hormone de croissance–IFG-1, atténuant la sécrétion de l’hormone de croissance. À long terme, cela est associé à une morbidité liée à la croissance chez les patients atteints de CE.37
En raison de cela, il y a eu un changement de paradigme dans la dérivation intestinale initiale et la prise en charge de l’intestin postérieur. Dans leur série de 77 patients, Sawaya et al ont constaté que la longueur de l’intestin postérieur variait largement de 2 à >20 cm, avec plus de la moitié dans l’intervalle de 6 à 15 cm. Fait intéressant, les patients avec un génotype XY étaient plus susceptibles de présenter une longueur de l’intestin postérieur inférieure à 10 cm comparativement à leurs homologues XX. Dans cette série, seulement 10 patients ont subi une résection de l’intestin postérieur, et uniquement en cas de viabilité douteuse de l’intestin postérieur ou de longueur extrêmement courte. En conséquence, les auteurs ont préconisé la tubulisation cæcale avec création d’une colostomie terminale afin d’éliminer le syndrome de l’intestin court et de faciliter les procédures d’abaissement intestinal.20 En général, la reconstruction gastro-intestinale est réalisée 1 à 2 ans après la dérivation fécale initiale; toutefois, si cette reconstruction est associée à la fermeture vésicale, le rapprochement du pubis est primordial pour la réussite de la reconstruction vésicale, de la paroi abdominale et intestinale. Habituellement, le succès de ces reconstructions repose sur une reconstruction pelvienne avec ostéotomies.17
Dans la même série, Sawaya et al ont réalisé huit procédures d’abaissement intestinal—7 avant l’âge de 5 ans, allant de 2 à 12 ans—et tous les patients avaient un intestin postérieur préservé d’au moins 10 cm de longueur.20 Parmi les autres facteurs à considérer pour décider de réaliser un abaissement intestinal figurent la capacité du patient à émettre des selles moulées et la mise en évidence d’une musculature sphinctérienne anale réactive à la stimulation lors de l’examen sous anesthésie. Chez les enfants qui ne sont pas candidats à un abaissement intestinal, une stomie fécale définitive est l’option la plus durable pour la prise en charge à long terme. À noter que, si le reliquat d’intestin postérieur n’est pas intégré au transit fécal, il doit être conservé en vue d’une augmentation vésicale ultérieure ou d’une reconstruction vaginale.17
S’il est établi, au stade initial de la fermeture de l’omphalocèle, qu’une fermeture concomitante de la vessie et de la paroi abdominale ne peut pas être réalisée de façon sûre et avec succès, les hémivessies doivent alors être rapprochées sur la ligne médiane, convertissant le défaut en une CBE.17 Cela permet la distension abdominale et l’agrandissement de la plaque vésicale en vue d’une fermeture différée ultérieure. Enfin, l’intestin postérieur est laissé à ce stade en fistule muqueuse s’il n’est pas incorporé à la reconstruction intestinale initiale.
Reconstruction urinaire
Réparation moderne en plusieurs temps
La reconstruction urinaire de la CE est similaire à celle de la CBE. Le rapprochement postérieur des hémivessies convertit la malformation de CE en CBE et implique la dissection des faces latérales des moitiés vésicales à partir de la paroi abdominale, ainsi que la fermeture sur la ligne médiane.38 La restauration de l’anatomie par la mise en place de la vessie et de l’urètre postérieur profondément dans le bassin demeure un facteur crucial pour la réussite de la reconstruction chirurgicale. Le rapprochement d’un bassin élargi contribue à cet objectif, en permettant la reconstruction de la paroi abdominale et des voies urinaires, toutefois, cela nécessite généralement des ostéotomies pelviennes et une fixation, comme cela sera décrit plus loin dans ce chapitre.
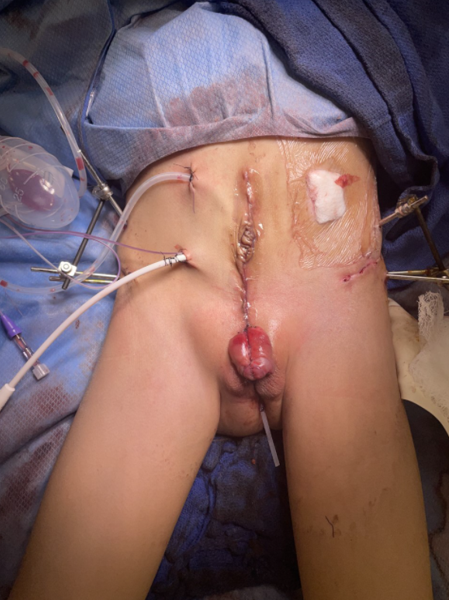
Figure 6 Réparation moderne en plusieurs temps de l’exstrophie. Photographie postopératoire du patient vu sur la photographie préopératoire de la Figure 3. Remarquer la mise en place des broches d’ostéotomie avant la pose du fixateur externe.
Le grand défaut de la paroi abdominale observé dans la CE représente un défi considérable pour la reconstruction de la paroi abdominale sans tension (Figure 7). Le fait de ne pas tenir compte de la tension au moment de la fermeture peut avoir un impact sur les taux de fermeture réussie et la formation de fistules. Les matériaux bioprothétiques tels que Alloderm (Allergan, Branchburg, NJ) se sont révélés très prometteurs pour pallier le manque de tissu de la paroi abdominale tout en assurant une fermeture sans tension.39 En outre, Alloderm a été utilisé comme adjuvant pour réduire la fistulisation pénopubienne par la couverture de la suture interpubienne au moment de la reconstruction des voies urinaires.40
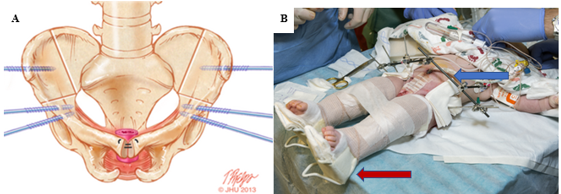
Figure 7 A, localisation des ostéotomies combinées bilatérales (innominée antérieure et iliaque verticale) marquées sur l’os, avec l’emplacement idéal des points d’insertion des broches. B, traction de Buck modifiée (flèche rouge) et fixateur externe (flèche bleue) à droite.
Pour assurer la concordance avec le genre assigné, la reconstruction des organes génitaux externes est réalisée dès que possible au cours de la période postnatale immédiate. Chez les mâles génotypiques, cela garantit qu’ils sont élevés en concordance avec leur phénotype. Alors que les effets psychosociaux et psychosexuels à long terme chez les enfants qui subissent une réassignation de genre font actuellement l’objet d’un grand intérêt, comme discuté précédemment, les études histologiques des testicules chez des sujets masculins ayant subi une réassignation de genre confirment une histologie normale, et ce malgré la cryptorchidie.41 Compte tenu du caractère souvent modeste et asymétrique du tissu pénien présent, la reconstruction phallique reste difficile, avec des résultats mitigés.
La phalloplastie s’est imposée comme une option de reconstruction efficace pour le remplacement pénien, bien que cela soit généralement différé jusqu’à la fin de l’adolescence (Figure 8).42 Cependant, dans les situations où l’insuffisance tissulaire impose une réassignation masculin-vers-féminin, la reconstruction génitale initiale doit réunir les hémiphalles sur la ligne médiane pour former un clitoris. Lorsque le tissu phallique est adéquat, toutefois, la réparation de l’épispadias est réalisée à l’âge de 1 an, souvent en utilisant la réparation de Cantwell-Ransley, bien décrite.43 Chez les sujets de sexe féminin génétique, la reconstruction vaginale est effectuée précocement, et chez les sujets de sexe masculin réassignés au féminin, une reconstruction différée d’un néovagin est appropriée, avec nécessité de dilatations au long cours du néovagin.44 En raison de la brièveté de l’urètre chez les sujets de sexe féminin génétique, la réparation de l’épispadias isolé est généralement associée à une reconstruction du col vésical selon Young-Dees-Leadbetter (BNR), une monoplastie et une clitoroplastie.
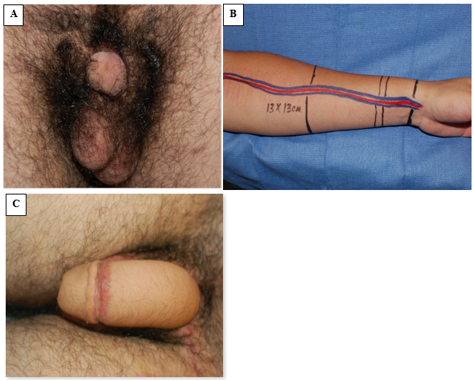
Figure 8 Phalloplastie par lambeau libre antébrachial radial. A, photographie préopératoire montrant un tissu pénien peu développé. B, préparation peropératoire du tissu du lambeau antébrachial et du pédicule vasculaire associé. C, photographie postopératoire montrant le résultat de la phalloplastie.
L’importance de l’ostéotomie
L’ostéotomie s’est imposée comme un facteur déterminant de la fermeture réussie chez la plupart des enfants atteints de CBE et elle est indiquée chez tous les enfants atteints de CE au moment de la fermeture vésicale.17,44 L’ostéotomie corrige le large diastasis pubien et permet un positionnement orthotopique de la vessie et de l’urètre postérieur aussi profondément que possible dans le bassin, tout en facilitant la reconstruction de la vessie et de la paroi abdominale avec une tension minimale. Cette réduction de tension est corrélée à une diminution des taux de déhiscence et d’éventrations postopératoires. De plus, il a été constaté que l’ostéotomie réduit la probabilité de complications significatives, passant de 89 % chez les patients dont la fermeture a été réalisée sans ostéotomie à 17 % chez les enfants dont la fermeture a été réalisée avec ostéotomie.45 Cela a été confirmé dans une grande série de 80 patients atteints de CE, qui a rapporté 91 % de réparations d’exstrophie réussies lorsque l’ostéotomie était réalisée au moment de la fermeture.46 Bien que l’ostéotomie ait contribué à une fermeture réussie, elle n’a pas montré d’impact sur la continence ultérieure chez les patients atteints de CE.
Les ostéotomies iliaques antérieures et verticales bilatérales combinées constituent la méthode privilégiée dans notre établissement et sont coordonnées avec la fermeture vésicale selon une approche appelée voie en deux temps (DSP).47 Cette approche de l’ostéotomie en décubitus dorsal présente l’avantage supplémentaire de ne pas nécessiter de repositionner le patient avant la fermeture de la vessie et de la paroi abdominale (Figure 7). Comme une voie d’abord postérieure est évitée, les risques d’endommager toute réparation rachidienne sont également minimisés. En outre, les ostéotomies pelviennes permettent une réduction pelvienne progressive avec une fixation externe 2 à 3 semaines avant la fermeture vésicale et de la paroi abdominale. Cela est souvent utilisé en cas de diastasis pubien extrêmement large (> 10 cm) et a été utilisé avec succès chez un patient présentant un diastasis pubien de 16 cm.47 Dans notre établissement, la fixation externe et la traction de Buck modifiée sont maintenues pendant 6 à 8 semaines afin d’assurer une cicatrisation adéquate. Selon notre expérience, cette technique d’immobilisation fournit des résultats exceptionnels avec un taux d’échec de 3,8 % lors des fermetures primaires, contre 65,7 % pour une immobilisation avec un plâtre pelvi-jambier.48
Reconstruction en un temps
Une autre approche de la réparation chez le patient atteint de CE est la réparation en un temps, ou réparation primaire complète de l’exstrophie (CPRE), popularisée par Grady et Mitchell.49 Contrairement à l’approche en plusieurs temps de reconstruction abdominale et vésicale, suivie d’une réparation de l’épispadias et d’une procédure de continence, la réparation en un temps combine des éléments de chaque étape en une seule réparation. La procédure est similaire à celle décrite pour la CBE, toutefois elle prend en compte le moment de la fermeture par rapport à l’omphalocèle, en mettant l’accent sur le report de la fermeture en cas d’omphalocèle volumineuse ou d’autres problèmes médicaux particuliers. D’autres ont conclu que, bien que la réparation en un temps soit applicable à un groupe restreint de patients, certains facteurs pouvant en contre-indiquer l’utilisation sont une petite plaque vésicale, un diastasis pubien >6 cm, ou d’autres affections médicales telles qu’un dysraphisme spinal, lesquelles, même en l’absence d’exstrophie, peuvent nécessiter de multiples interventions chirurgicales .50 Dans une série limitée de 6 patients, Lee et al ont rapporté une fermeture réussie avec une réparation primaire complète. Ils ont toutefois noté la formation d’un septum vésical médian avec hydronéphrose associée chez deux enfants. En outre, un enfant avait une miction spontanée et un autre a subi une cystoplastie d’augmentation (AC).51
Les partisans de cette technique soutiennent qu’elle peut réduire le coût, la morbidité et le traumatisme associés à des interventions multiples, et même stimuler la croissance précoce de la vessie. La réparation de l’épispadias, dans ce cas, est réalisée par démontage pénien, au cours duquel la plaque urétrale est entièrement disséquée des corps caverneux. Cela permet la fermeture urétrale et place le col vésical en position postérieure dans le bassin.52 Les partisans du CPRE attribuent la diminution du nombre d’opérations nécessaires pour obtenir la continence à une augmentation de la résistance de la voie de sortie vésicale à un âge précoce. Cependant, de nombreux patients nécessitent encore une BNR et, notamment, seuls 20-56% des patients CPRE qui restent incontinents obtiennent la continence après une BNR supplémentaire.53,54
Complications
Les réparations en un temps et en plusieurs temps sont des techniques de reconstruction acceptables dans la CE. Bien que chaque approche ait ses mérites, les résultats rapportés situent le taux d’échec d’une reconstruction en un temps à 48 % contre 15 % pour une réparation en plusieurs temps. La désunion de plaie, le prolapsus vésical, l’obstruction de la sortie vésicale et la formation d’une fistule vésico-cutanée doivent toutes être considérées comme des échecs de fermeture et ont été rapportées tant avec les techniques en un temps qu’avec les techniques en plusieurs temps.
Quelle que soit l’approche chirurgicale, la réussite de la fermeture vésicale primaire est l’objectif principal. Un échec de fermeture est dévastateur pour le patient et sa famille, et les répercussions sont importantes. Les patients présentant une CE et connaissant un échec de fermeture primaire sont exposés à davantage d’interventions et à une exposition accrue à l’anesthésie générale. Bien qu’un échec de fermeture vésicale dans la CBE ait un impact négatif sur la continence urinaire finale, cet impact spécifique sur la continence est moins marqué chez les patients atteints de CE, car la plupart de ces patients nécessitent des procédures supplémentaires de continence, quel que soit le résultat primaire.55,56 Les répercussions vont au-delà des interventions supplémentaires et incluent une morbidité financière. Goldstein et al ont réalisé une analyse des coûts chez des patients atteints de CE et ont constaté qu’en cas de réparation primaire réussie de la CE, il faut environ $196,000 pour atteindre la continence, tandis que le coût pour obtenir la continence s’élève à $407,000 après un échec de la fermeture vésicale primaire.57
Une large série évaluant les facteurs prédictifs d’une fermeture primaire réussie dans l’exstrophie a rapporté qu’une fermeture de la CE en plusieurs étapes, ainsi que le recours à l’ostéotomie pelvienne, constituaient des facteurs prédictifs indépendants significatifs d’une fermeture réussie. De plus, après la fermeture vésicale, l’immobilisation postopératoire était cruciale pour le succès.58 Pour cette raison, nous recommandons l’utilisation d’une fixation externe combinée à une traction, comme c’est la norme dans notre établissement. D’autres méthodes, telles que les plâtres pelvi-jambiers (spica), peuvent ne pas assurer une immobilisation adéquate et peuvent donc entraîner une augmentation des échecs de la reconstruction pelvienne. Les complications de l’ostéotomie sont rares et souvent autolimitantes, mais comprennent un risque accru de parésies nerveuses et musculaires transitoires (se résolvent généralement dans les 12 semaines suivant la chirurgie), un retard de consolidation iliaque, et une infection ou inflammation superficielles au point d’entrée des broches.59 Dans l’expérience de l’établissement des auteurs, bien que l’ostéotomie pelvienne avec immobilisation postopératoire puisse prolonger la durée d’hospitalisation, la diminution drastique du taux d’échec est jugée bénéfique par les patients et les soignants.
Des données émergentes suggèrent que le moment de la fermeture initiale influence les taux de réussite. En 2014, Shah et ses collègues ont rapporté les résultats chez 60 patients atteints de CE et ont constaté qu’une augmentation de 1 cm du diastasis pré-fermeture entraînait une augmentation par 2.64 des chances d’échec de la fermeture initiale. La réalisation secondaire d’une ostéotomie et le report de la fermeture, tout en permettant au fixateur externe de diminuer le diastasis, augmentaient le taux de réussite de la fermeture. Fait intéressant, 77% des patients ayant présenté un échec de fermeture avaient été fermés au cours de la première semaine de vie, contre 26% des patients ayant bénéficié d’une fermeture étagée. En outre, seulement 31% des patients du groupe avec échec de fermeture ont subi une ostéotomie lors de la fermeture initiale, contre 82% des patients avec fermeture étagée. Parmi les autres stratégies protectrices contre l’échec figure le report de la fermeture initiale de la vessie jusqu’après la fermeture de l’omphalocèle.60 Enfin, 92% des patients ayant bénéficié d’une re-fermeture réussie ont subi une ostéotomie.47 Ces résultats, démontrant une augmentation du succès avec une réparation primaire différée, ont été étayés par une autre série qui a montré un pourcentage plus élevé d’échecs de la fermeture vésicale primaire chez les patients chez lesquels la fermeture a été réalisée dans les 30 premiers jours de vie par rapport à ceux opérés après 30 jours d’âge (47.7% vs 19.3%).55
Pour obtenir la continence après un échec de la fermeture primaire, il faut réaliser une fermeture vésicale secondaire. À l’instar des résultats rapportés par Shah et al., Davis et ses collègues ont constaté que l’âge auquel la fermeture vésicale secondaire est réalisée est prédictif du succès global. Ils ont rapporté que les patients ayant bénéficié d’une fermeture vésicale secondaire réussie étaient de plus d’un an plus âgés que les patients chez qui la fermeture vésicale secondaire avait échoué (âge médian de 104 semaines contre 38 semaines).56 La fermeture secondaire réussie présentait un délai médian de 154 semaines, contre un délai médian de 30 semaines pour les fermetures secondaires ayant échoué. L’échec de la reconstruction urinaire chez les enfants atteints d’exstrophie vésicale est significatif, car il peut entraîner un ralentissement de la croissance vésicale et une perte du patron vésical, ce qui peut rendre l’obtention de la continence de plus en plus difficile, voire impossible.
Acquisition de la continence
En raison de la conjonction fréquente d’anomalies possibles — petites plaques vésicales, vessies peu complaisantes, col vésical largement béant (s’il est présent) et anomalies rachidiennes contribuant à un intestin et une vessie neurogènes — la continence mictionnelle est peu probable. Cependant, la continence demeure l’objectif ultime dans la reconstruction de l’exstrophie afin de permettre à ces enfants de mener une vie plus proche de la normale. À cette fin, des interventions supplémentaires sont souvent nécessaires, comme l’ont confirmé Maruf et al dans une vaste série rapportant que les patients nécessitent une médiane de 2 (plage 1–4) interventions de continence urinaire pour obtenir un intervalle sec supérieur à 3 heures.61
Dans une grande série provenant de l’établissement des auteurs, évaluant les résultats chez 80 patients atteints du complexe OEIS, des données sur la reconstruction urinaire étaient disponibles pour 73 patients. Parmi les 40 patients continents, 38 ont conservé leur vessie native, tandis que 2 avaient des néovessies issues de l’intestin grêle (poche de Koch). Parmi les 38 patients porteurs d’une vessie native, 30 (79 %) ont reçu un conduit cathétérisable continent (dérivation urinaire continente, CUD) utilisant soit l’appendice (n =3), soit un segment intestinal (n = 27), et 32 (84 %) ont également bénéficié d’une cystoplastie d’augmentation (AC) avant d’obtenir la continence. Seuls 9 patients (24 %) ont conservé un urètre perméable et continent.46 Dans un ensemble d’études évaluant la continence urinaire dans le CE, une AC a été nécessaire chez jusqu’à 50 % des patients, avec un taux global de continence de 72 %.62
Ces interventions complémentaires pour la continence, AC et CUD, présentent des complications spécifiques, notamment une surproduction de mucus, des calculs vésicaux, une colonisation bactérienne chronique et des polypes épithéliaux. D’autres problèmes à long terme découlent de l’AC, notamment une acidose métabolique chronique et, plus rarement, un carcinome.63,64 Avec le temps, la stomie créée pour CUD peut se sténoser, prolaber, nécroser ou fuir, nécessitant une révision.64
Chez les patientes génétiquement de sexe féminin, une continence satisfaisante peut être obtenue après une BNR de Young-Dees-Leadbetter, bien que la plupart des patientes nécessitent un cathétérisme intermittent propre. Les techniques utilisées chez les patients des deux sexes pour augmenter la capacité vésicale ont été décrites précédemment, mais comprennent l’incorporation d’un segment de l’intestin postérieur (lorsqu’il est disponible), de l’intestin grêle et du côlon, et, plus rarement, de l’estomac.38 Obtenir la continence est beaucoup plus difficile chez la patiente réassignée de masculin vers féminin et une stomie continente peut être la stratégie la plus avantageuse à long terme.17 Quelle que soit la technique employée, d’après l’expérience de notre établissement, les enfants sont continents pour les urines à un âge médian de 11 ans.61 Dans une grande série évaluant la continence chez des patients atteints d’exstrophie cloacale, 50 % (35 sur 61) des enfants ont atteint la continence, dont 30 sur 35 pratiquaient un cathétérisme intermittent via une stomie continente et les autres urinaient spontanément ou se cathétérisaient par l’urètre.65
Suivi recommandé
Les patients atteints d’exstrophie, leurs familles, et les équipes médicales et chirurgicales qui les prennent en charge développent un lien particulier, unique en médecine. Après la reconstruction des voies urinaires et de la paroi abdominale et l’éventuelle réparation ultérieure de l’épispadias, l’objectif de la prise en charge s’oriente vers la protection des voies urinaires supérieures. À cette fin, dans l’établissement de l’auteur, nous surveillons tout signe de reflux au moyen de cystographies et de cystoscopies annuelles, tout en suivant la croissance vésicale. Chez les enfants d’âge scolaire (au moins 5 à 7 ans), nous évaluons la capacité vésicale, réalisons un bilan urodynamique, et apprécions la capacité de l’enfant à participer à ses propres soins, par l’évaluation de sa dextérité et de sa maturité, avant la reconstruction du col vésical.31
Conclusions
Le CE est une malformation congénitale multisystémique dévastatrice qui, autrefois, prédisposait les enfants à une morbidité importante et même à la mortalité. Des progrès considérables, cependant, dans la prise en charge chirurgicale et médicale de ces enfants leur ont permis de mener une vie quasi normale jusqu’à l’âge adulte. Le succès de la prise en charge contemporaine découle d’une approche multidisciplinaire visant à surmonter les défis auxquels ces patients sont confrontés. À mesure que ces patients continuent de mener une vie saine tout au long de l’âge adulte, nous continuerons d’en apprendre davantage en ce qui concerne la qualité de vie et les moyens d’optimiser la fonction globale des patients.
Points clés
- Un diagnostic périnatal précis et approfondi est crucial pour organiser une prise en charge multidisciplinaire appropriée—fUS et fMRI sont des modalités utiles dans cette démarche.
- La prise en charge postnatale initiale doit garantir la stabilité médicale du patient. Une fois stable, un examen clinique approfondi et des examens d’imagerie appropriés doivent être réalisés afin d’assurer l’identification complète de toutes les anomalies associées.
- La reconstruction abdominale et des voies urinaires ne peut avoir lieu qu’après une évaluation neurochirurgicale de l’atteinte rachidienne ou du SNC et l’obtention de l’aval.
- Tout reliquat d’intestin postérieur doit être préservé à tout prix chez ces enfants, car il peut être utilisé pour la reconstruction gastro-intestinale ou génito-urinaire.
- La séparation de la vessie du tractus gastro-intestinal avec dérivation fécale concomitante est essentielle avant toute reconstruction ultérieure. La colostomie terminale s’est imposée comme la forme de dérivation fécale la moins morbide.
- Un suivi rapproché après la reconstruction chez ces patients est essentiel afin d’assurer la préservation de la fonction des voies urinaires supérieures.
Perspectives futures
Les recherches actuelles visent à améliorer l’accompagnement et à réduire la morbidité associée à la chirurgie en développant de meilleurs outils prédictifs pour les patients et leurs familles. Un meilleur accompagnement peut être obtenu grâce à des outils, tels que ceux qui prédisent la capacité et la croissance vésicales (Sholklapper et al 2022, soumis), ce qui peut avoir des implications sur les interventions chirurgicales futures et la capacité à atteindre la continence.66 De plus, des recherches fondamentales évaluant les différences d’ultrastructure entre les vessies atteintes d’exstrophie vésicale et des témoins appariés selon l’âge pourraient fournir une image plus claire des courbes de croissance vésicale différentielles chez ces patients. Le recours à l’imagerie pour évaluer l’anatomie du plancher pelvien, en tant qu’outil de planification chirurgicale, apporte des informations supplémentaires pour la prise de décision peropératoire, et l’utilisation de cette imagerie devrait s’étendre. À long terme, aborder le champ en expansion de l’urologie translationnelle et la question de qui assurera la prise en charge de ces enfants lorsqu’ils entreront dans l’âge adulte aidera à optimiser les soins de ces patients. Enfin, l’essor de la télémédecine pour les consultations entre centres médicaux pourrait améliorer la communication au sujet de cette affection rare et améliorer le bien-être des patients et des familles en réduisant la charge liée à l’accessibilité, aux déplacements et aux coûts.
Lectures recommandées
- Gearhart JP, Carlo HN. Exstrophy-epispadias complex. 12th ed., Philadelphia, PA: Elsevier; 2020, DOI: 10.1201/9780429194993-25.
- Grady RW, Mitchell ME. Complete Primary Repair Of Exstrophy. J Urol 1999; 65 (1): 1415–1420. DOI: 10.1097/00005392-199910000-00071.
- Davis R, Stewart D, Maruf M, Lau H, Gearhart JP. Complex abdominal wall reconstruction combined with bladder closure in OEIS complex. J Pediatr Surg 2019; 54 (11): 2408–2412. DOI: 10.1016/j.jpedsurg.2019.03.022.
- Tourchi A, Inouye BM, Di Carlo HN, Young E, Ko J, Gearhart JP. New advances in the pathophysiologic and radiologic basis of the exstrophy spectrum. Journal of Pediatric Urology 2014; 10 (2): 212–218.
- Kasprenski M, Michaud J, Yang Z, Maruf M, Benz K, Jayman J, et al.. Urothelial differences in the exstrophy-epispadias complex: potential implications for management. The Journal of Urology 2021; 205 (5): 1460–1465.
- Di Carlo HN, Maruf M, Massanyi EZ, Shah B, Tekes A, Gearhart JP. 3-dimensional magnetic resonance imaging guided pelvic floor dissection for bladder exstrophy: a single arm trial. The Journal of Urology 2019; 202 (2): 406–412.
Références
- Muecke EC. The Role of the Cloacae Membrane in Exstrophy: The First Successful Experimental Study. J Urol 1069; 92 (6): 659–668. DOI: 10.1016/s0022-5347(17)64028-x.
- Woo LL, Thomas JC, Brock JW. Cloacal exstrophy: A comprehensive review of an uncommon problem. J Pediatr Urol 2010; 6 (2): 102–111. DOI: 10.1016/j.jpurol.2009.09.011.
- Moore KL. Organogenetic Period: The Fourth to Eighth Weeks. In: Moore KL, Persaud TVN, editors. The Developing Human: Clinically Oriented Embryology. 7th ed. Philadelphia, PA: Saunders; 2003.
- Howell C, Caldamone A, Snyder H, Ziegler M, Duckett J. Optimal management of cloacal exstrophy. J Pediatr Surg 1983; 18 (4): 365–369. DOI: 10.1016/s0022-3468(83)80182-1.
- Cervellione RM, Mantovani A, Gearhart J, Bogaert G, Gobet R, Caione P, et al.. Prospective study on the incidence of bladder/cloacal exstrophy and epispadias in Europe. J Pediatr Urol 2015; 11 (6): 337.e1–337.e6. DOI: 10.1016/j.jpurol.2015.03.023.
- Shapiro E, Lepor H, Jeffs RD. The Inheritance of the Exstrophy-Epispadias Complex. J Urol 1984; 132 (2): 308–310. DOI: 10.1016/s0022-5347(17)49605-4.
- Ives E, Coffey R, Carter CO. A family study of bladder exstrophy. J Med Genet 1980; 17 (2): 139–141. DOI: 10.1136/jmg.17.2.139.
- Birth Defects Monitoring Systems IC for. Epidemiology of bladder exstrophy and epispadias: A communication from the international clearinghouse for birth defects monitoring systems. Teratology 1987; 36 (2): 221–227. DOI: 10.1002/tera.1420360210.
- Botto LD, Feldkamp ML, Amar E, Carey JC, Castilla EE, Clementi M, et al.. Acardia: Epidemiologic findings and literature review from the International Clearinghouse for Birth Defects Surveillance and Research. Am J Med Genet C Semin Med Genet 2011; 157 (4): 262–273. DOI: 10.1002/ajmg.c.30318.
- Boyadjiev SA, Dodson JL, Radford CL, Ashrafi GH, Beaty TH, Mathews RI, et al.. Clinical and molecular characterization of the bladder exstrophy-epispadias complex: analysis of 232 families. BJU Int 2004; 94 (9): 1337–1343. DOI: 10.1111/j.1464-410x.2004.05170.x.
- WOOD HADLEYM, TROCK BRUCEJ, GEARHART JOHNP. In Vitro Fertilization and the Cloacal-Bladder Exstrophy-Epispadias Complex: Is there an Association? J Urol 2003; 169 (4): 1512–1515. DOI: 10.1097/01.ju.0000054984.76384.66.
- Reutter H, Boyadjiev SA, Gambhir L, Ebert A-K, Rösch WH, Stein R, et al.. Phenotype Severity in the Bladder Exstrophy-Epispadias Complex: Analysis of Genetic and Nongenetic Contributing Factors in 441 Families from North America and Europe. J Pediatr 2011; 159 (5): 825–831.e1. DOI: 10.1016/j.jpeds.2011.04.042.
- Sponseller PD, Bisson LJ, Gearhart JP, Jeffs RD, Magid D, Fishman E. The anatomy of the pelvis in the exstrophy complex. J Bone Joint Surg Am 1995; 77 (2): 177–189. DOI: 10.2106/00004623-199502000-00003.
- Stec AA, Pannu HK, Tadros YE, Sponseller PD, Fishman EK, Gearhart JP. Pelvic Floor Anatomy In Classic Bladder Exstrophy Using 3-dimensional Computerized Tomography: J Urol 2001; 166 (4): 1444–1449. DOI: 10.1097/00005392-200110000-00066.
- Greene WB, Dias LS, Lindseth RE, Torch MA. Musculoskeletal problems in association with cloacal exstrophy. J Bone Joint Surg Am 1991; 73 (4): 551–560. DOI: 10.2106/00004623-199173040-00012.
- Davidoff AM, Hebra A, Balmer D, Templeton JM, Schnaufer L. Management of the gastrointestinal tract and nutrition in patients with cloacal exstrophy. J Pediatr Surg 1996; 31 (6): 771–773. DOI: 10.1016/s0022-3468(96)90129-3.
- Mathews R, Jeffs RD, Reiner WG, Docimo SG, Gearhart JP. Cloacal Exstrophy-improving The Quality Of Life. J Urol 1998; 6 (2): 2452–2456. DOI: 10.1097/00005392-199812020-00017.
- McHoney M, Ransley PG, Duffy P, Wilcox DT, Spitz L. Cloacal exstrophy: Morbidity associated with abnormalities of the gastrointestinal tract and spine. J Pediatr Surg 2004; 39 (8): 1209–1213. DOI: 10.1016/j.jpedsurg.2004.04.019.
- Sugar EC, Firlit CF. Management of cloacal exstrophy. Urology 2004; 32 (4): 320–322. DOI: 10.1016/0090-4295(88)90234-8.
- Sawaya D, Goldstein S, Seetharamaiah R, Suson K, Nabaweesi R, Colombani P, et al.. Gastrointestinal ramifications of the cloacal exstrophy complex: a 44-year experience. J Pediatr Surg 2010; 45 (1): 171–176. DOI: 10.1016/j.jpedsurg.2009.10.030.
- Hurwitz RS, Manzoni GAM, Ransley PG, Stephens FD. Cloacal Exstrophy: A Report of 34 Cases. J Urol 1987; 138 (4 Part 2): 1060–1064. DOI: 10.1016/s0022-5347(17)43502-6.
- Phillips TM, Salmasi AH, Stec A, Novak T, Gearhart JP, Mathew J. Re: Urological Outcomes in the Omphalocele Exstrophy Imperforate Anus Spinal Defects (OEIS) Complex: Experience with 80 Patients. J Urol 2013; 191 (4): 1118–1119. DOI: 10.1016/j.juro.2014.01.044.
- Schlegel PN, Gearhart JP. Neuroanatomy of the Pelvis in an Infant with Cloacal Exstrophy: A Detailed Microdissection with Histology. J Urol 1989; 141 (3 Part 1): 583–585. DOI: 10.1016/s0022-5347(17)40901-3.
- Connolly JA, Peppas DS, Jeffs RD, Gearhart JP. Prevalence and Repair of Inguinal Hernias in Children with Bladder Exstrophy. J Urol 1995; 154 (5): 1900–1901. DOI: 10.1097/00005392-199511000-00093.
- Sadula SR, Kanhere SV, Phadke VD. Exstrophy of Cloaca Sequence (Oeis Complex) With Multiple Cardiac Malformations. Indian Journal of Case Reports 2019; 05 (04): 317–319. DOI: 10.32677/ijcr.2019.v05.i04.006.
- Meizner I, Bar-Ziv J. In utero prenatal ultrasonic diagnosis of a rare case of cloacal exstrophy. J Clin Ultrasound 1985; 13 (7): 500–502. DOI: 10.1002/jcu.1870130714.
- Austin PF, Homsy YL, Gearhart JP, Porter K, Guidi C, Madsen K, et al.. The Prenatal Diagnosis Of Cloacal Exstrophy. J Urol 1998; 160 (3.2): 1179–1181. DOI: 10.1097/00005392-199809020-00061.
- Calvo-Garcia MA, Kline-Fath BM, Rubio EI, Merrow AC, Guimaraes CV, Lim F-Y. Fetal MRI of cloacal exstrophy. Pediatr Radiol 2013; 43 (5): 593–604. DOI: 10.1007/s00247-012-2571-3.
- Goto S, Suzumori N, Obayashi S, Mizutani E, Hayashi Y, Sugiura-Ogasawara M. Prenatal findings of omphalocele-exstrophy of the bladder-imperforate anus-spinal defects (OEIS) complex. Congenit Anom (Kyoto) 2012; 52 (3): 179–181. DOI: 10.1111/j.1741-4520.2011.00342.x.
- Weiss DA, Oliver ER, Borer JG, Kryger JV, Roth EB, Groth TW, et al.. Key anatomic findings on fetal ultrasound and MRI in the prenatal diagnosis of bladder and cloacal exstrophy. J Pediatr Urol 2020; 16 (5): 665–671. DOI: 10.1016/j.jpurol.2020.07.024.
- Lowentritt BH, Van Zijl PS, Frimberger D, Baird A, Lakshmanan Y, Gearhart JP. Variants Of The Exstrophy Complex: A Single Institution Experience. J Urol 2005; 173 (5): 1732–1737. DOI: 10.1097/01.ju.0000154353.03056.5c.
- Gearhart JP, Jeffs RD. The bladder exstrophy-epispadias complex. 3rd ed., Philadelphia, PA: Saunders; 1998, DOI: 10.1007/978-3-319-44182-5_13.
- Tank ES, Lindenauer SM. Principles of management of exstrophy of the cloaca. Am J Surg 1970; 119 (1): 95–98. DOI: 10.1016/0002-9610(70)90018-8.
- Reiner WG, Gearhart JP. Discordant Sexual Identity in Some Genetic Males with Cloacal Exstrophy Assigned to Female Sex at Birth. N Engl J Med 2004; 350 (4): 333–341. DOI: 10.1056/nejmoa022236.
- Baker Towell DM, Towell AD. A Preliminary Investigation Into Quality of Life, Psychological Distress and Social Competence in Children With Cloacal Exstrophy. J Urol 2003; 169 (5): 1850–1853. DOI: 10.1097/01.ju.0000062480.01456.34.
- Mirheydar H, Evason K, Coakley F, Baskin LS, DiSandro M. 46, XY female with cloacal exstrophy and masculinization at puberty. J Pediatr Urol 2009; 5 (5): 408–411. DOI: 10.1016/j.jpurol.2009.03.013.
- Diamond DA, Burns JP, Mitchell C, Lamb K, Kartashov AI, Retik AB. Sex assignment for newborns with ambiguous genitalia and exposure to fetal testosterone: Attitudes and practices of pediatric urologists. J Pediatr 2006; 148 (4): 445–449. DOI: 10.1016/j.jpeds.2005.10.043.
- Fullerton BS, Sparks EA, Hall AM, Chan Y-M, Duggan C, Lund DP, et al.. Growth morbidity in patients with cloacal exstrophy: a 42-year experience. J Pediatr Surg 2016; 51 (6): 1017–1021. DOI: 10.1016/j.jpedsurg.2016.02.075.
- Gearhart JP, Jeffs RD. Techniques to Create Urinary Continence in the Cloacal Exstrophy Patient. J Urol 1991; 146 (2 Part 2): 616–618. DOI: 10.1016/s0022-5347(17)37871-0.
- Davis R, Stewart D, Maruf M, Lau H, Gearhart JP. Complex abdominal wall reconstruction combined with bladder closure in OEIS complex. J Pediatr Surg 2019; 54 (11): 2408–2412. DOI: 10.1016/j.jpedsurg.2019.03.022.
- Henderson CG, North AC, Gearhart JP. The use of alloderm as an adjunct in the closure of the bladder – Cloacal exstrophy complex. J Pediatr Urol 2011; 7 (1): 44–47. DOI: 10.1016/j.jpurol.2010.02.209.
- Mathews, Perlman, Marsh, Gearhart. Gonadal morphology in cloacal exstrophy: implications in gender assignment. BJU Int 1999; 84 (1): 99–100. DOI: 10.1046/j.1464-410x.1999.00148.x.
- Lumen N, Monstrey S, Selvaggi G, Ceulemans P, De Cuypere G, Van Laecke E, et al.. Phalloplasty: A Valuable Treatment for Males with Penile Insufficiency. Urology 2008; 71 (2): 272–276. DOI: 10.1016/j.urology.2007.08.066.
- Gearhart JP, Leonard MP, Burgers JK, Jeffs RD. The Cantwell-Ransley Technique for Repair of Epispadias. J Urol 1992; 148 (3 Part 1): 851–854. DOI: 10.1016/s0022-5347(17)36742-3.
- Gearhart JP, Carlo HN. Exstrophy-epispadias complex. 12th ed., Philadelphia, PA: Elsevier; 2020, DOI: 10.1201/9780429194993-25.
- Ben-Chaim J, Peppas DS, Sponseller PD, Jeffs RD, Gearhart JP. Applications of Osteotomy in the Cloacal Exstrophy Patient. J Urol 1995; 154 (2.2): 865–867. DOI: 10.1097/00005392-199508000-00146.
- Mathews R, Gearhart JP, Bhatnagar R, Sponseller P. Staged Pelvic Closure of Extreme Pubic Diastasis in the Exstrophy-Epispadias Complex. J Urol 2006; 176 (5): 2196–2198. DOI: 10.1016/j.juro.2006.07.058.
- Benz KS, Jayman J, Maruf M, Baumgartner T, Kasprenski MC, Friedlander DA, et al.. Pelvic and lower extremity immobilization for cloacal exstrophy bladder and abdominal closure in neonates and older children. J Pediatr Surg 2018; 53 (11): 2160–2163. DOI: 10.1016/j.jpedsurg.2017.11.066.
- Grady RW, Mitchell ME. Complete Primary Repair Of Exstrophy. J Urol 1999; 65 (1): 1415–1420. DOI: 10.1097/00005392-199910000-00071.
- Thomas JC, DeMarco RT, Pope JC, Adams MC, Brock JW. First Stage Approximation of the Exstrophic Bladder in Patients With Cloacal Exstrophy–Should This be the Initial Surgical Approach in all Patients? J Urol 2007; 178 (4s): 1632–1636. DOI: 10.1016/j.juro.2007.03.164.
- Lee RS, Grady R, Joyner B, Casale P, Mitchell M. Can a Complete Primary Repair Approach be Applied to Cloacal Exstrophy? J Urol 2006; 176 (6): 2643–2648. DOI: 10.1016/j.juro.2006.08.052.
- Mitchell ME, Bagli DJ. Complete Penile Disassembly for Epispadias Repair. J Urol 1996; 155 (1): 300–304. DOI: 10.1097/00005392-199601000-00128.
- Gargollo P, Hendren WH, Diamond DA, Pennison M, Grant R, Rosoklija I, et al.. Bladder Neck Reconstruction is Often Necessary After Complete Primary Repair of Exstrophy. J Urol 2011; 185 (6s): 2563–2571. DOI: 10.1016/j.juro.2011.01.024.
- Schaeffer AJ, Stec AA, Purves JT, Cervellione RM, Nelson CP, Gearhart JP. Complete Primary Repair of Bladder Exstrophy: A Single Institution Referral Experience. J Urol 2011; 186 (3): 1041–1047. DOI: 10.1016/j.juro.2011.04.099.
- Friedlander DA, Di Carlo HN, Sponseller PD, Gearhart JP. Complications of bladder closure in cloacal exstrophy: Do osteotomy and reoperative closure factor in? J Pediatr Surg 2017; 52 (11): 1836–1841. DOI: 10.1016/j.jpedsurg.2016.12.002.
- Davis R, Sood A, Maruf M, Singh P, Kasprenski MC, DiCarlo HN, et al.. The failed bladder closure in cloacal exstrophy: Management and outcomes. J Pediatr Surg 2019; 54 (11): 2416–2420. DOI: 10.1016/j.jpedsurg.2019.02.012.
- Goldstein SD, Inouye BM, Reddy S, Lue K, Young EE, Abdelwahab M, et al.. Continence in the cloacal exstrophy patient: What does it cost? J Pediatr Surg 2016; 51 (4): 622–625. DOI: 10.1016/j.jpedsurg.2015.12.003.
- Jayman J, Tourchi A, Feng Z, Trock BJ, Maruf M, Benz K, et al.. Predictors of a successful primary bladder closure in cloacal exstrophy: A multivariable analysis. J Pediatr Surg 2019; 54 (3): 491–494. DOI: 10.1016/j.jpedsurg.2018.06.030.
- Inouye BM, Tourchi A, Di Carlo HN, Young EE, Gearhart JP. Modern Management of the Exstrophy-Epispadias Complex. Surg Res Pract 2014; 2014 (587064): 1–9. DOI: 10.1155/2014/587064.
- Shah BB, Di Carlo H, Goldstein SD, Pierorazio PM, Inouye BM, Massanyi EZ, et al.. Initial bladder closure of the cloacal exstrophy complex: Outcome related risk factors and keys to success. J Pediatr Surg 2014; 49 (6): 1036–1040. DOI: 10.1016/j.jpedsurg.2014.01.047.
- Maruf M, Kasprenski M, Jayman J, Goldstein SD, Benz K, Baumgartner T, et al.. Achieving urinary continence in cloacal exstrophy: The surgical cost. J Pediatr Surg 2018; 53 (10): 1937–1941. DOI: 10.1016/j.jpedsurg.2018.02.055.
- Mathews R. Achieving urinary continence in cloacal exstrophy. Semin Pediatr Surg 2011; 20 (2): 126–129. DOI: 10.1053/j.sempedsurg.2010.12.009.
- Surer I, Ferrer FA, Baker LA, Gearhart JP. Continent Urinary Diversion and the Exstrophy-Epispadias Complex. J Urol 2003; 169 (3): 1102–1105. DOI: 10.1097/01.ju.0000044921.19074.d0.
- Woodhouse CRJ, North AC, Gearhart JP. Standing the test of time: long-term outcome of reconstruction of the exstrophy bladder. World J Urol 2006; 24 (3): 244–249. DOI: 10.1007/s00345-006-0053-7.
- Suson K, Novak T, Gupta A, Benson J, Sponseller PD, Gearhart JP. The Neuro-Orthopedic Manifestations of the Omphalocele Exstrophy Imperforate Anus Spinal Defects (OEIS) Complex. J Pediatr Urol 2010; 5 (4): S54. DOI: 10.1016/j.jpurol.2009.02.083.
- Di Carlo HN, Maruf M, Massanyi EZ, Shah B, Tekes A, Gearhart JP. 3-dimensional magnetic resonance imaging guided pelvic floor dissection for bladder exstrophy: a single arm trial. The Journal of Urology 2019; 202 (2): 406–412.
- Tourchi A, Inouye BM, Di Carlo HN, Young E, Ko J, Gearhart JP. New advances in the pathophysiologic and radiologic basis of the exstrophy spectrum. Journal of Pediatric Urology 2014; 10 (2): 212–218.
- Kasprenski M, Michaud J, Yang Z, Maruf M, Benz K, Jayman J, et al.. Urothelial differences in the exstrophy-epispadias complex: potential implications for management. The Journal of Urology 2021; 205 (5): 1460–1465.
Dernière mise à jour: 2025-09-22 07:59