
12: Anomalies congénitales du rein et des voies urinaires
Ce chapitre prendra environ 15 minutes de lecture.
Introduction
Les anomalies congénitales du rein et des voies urinaires (CAKUT) constituent un groupe d’anomalies structurelles qui affectent les reins et les voies urinaires. On estime qu’il existe environ 4 à 60 cas pour 10 000 naissances dans la population générale. Le diagnostic de CAKUT est généralement posé lors d’une échographie prénatale de routine ou en postnatal chez un nouveau-né présentant des signes et symptômes cliniques associés.1 La prévalence des CAKUT chez les nouveau-nés prématurés était de 2 % dans une grande cohorte récente rapportée comprenant 409704 nourrissons.2
L’anomalie la plus fréquente est l’obstruction de la jonction pyélo-urétérale, qui touche environ 20 % des personnes atteintes de cette affection. Parmi les autres types de CAKUT, on retrouve les reins multikystiques dysplasiques, l’agénésie rénale, la dysplasie rénale, l’hypoplasie rénale, le reflux vésico-urétéral, le méga-uretère, la duplicité pyélo-urétérale, l’uretère ectopique et les valves de l’urètre postérieur (Figure 1).
Malgré les progrès significatifs réalisés dans la détection et le diagnostic du CAKUT, les causes sous-jacentes de cette affection ne sont pas encore entièrement comprises. Des recherches supplémentaires sont nécessaires pour améliorer notre compréhension des facteurs embryologiques, génétiques et environnementaux qui contribuent au développement du CAKUT et pour mieux comprendre leurs causes sous-jacentes et améliorer les stratégies de prévention et de traitement.
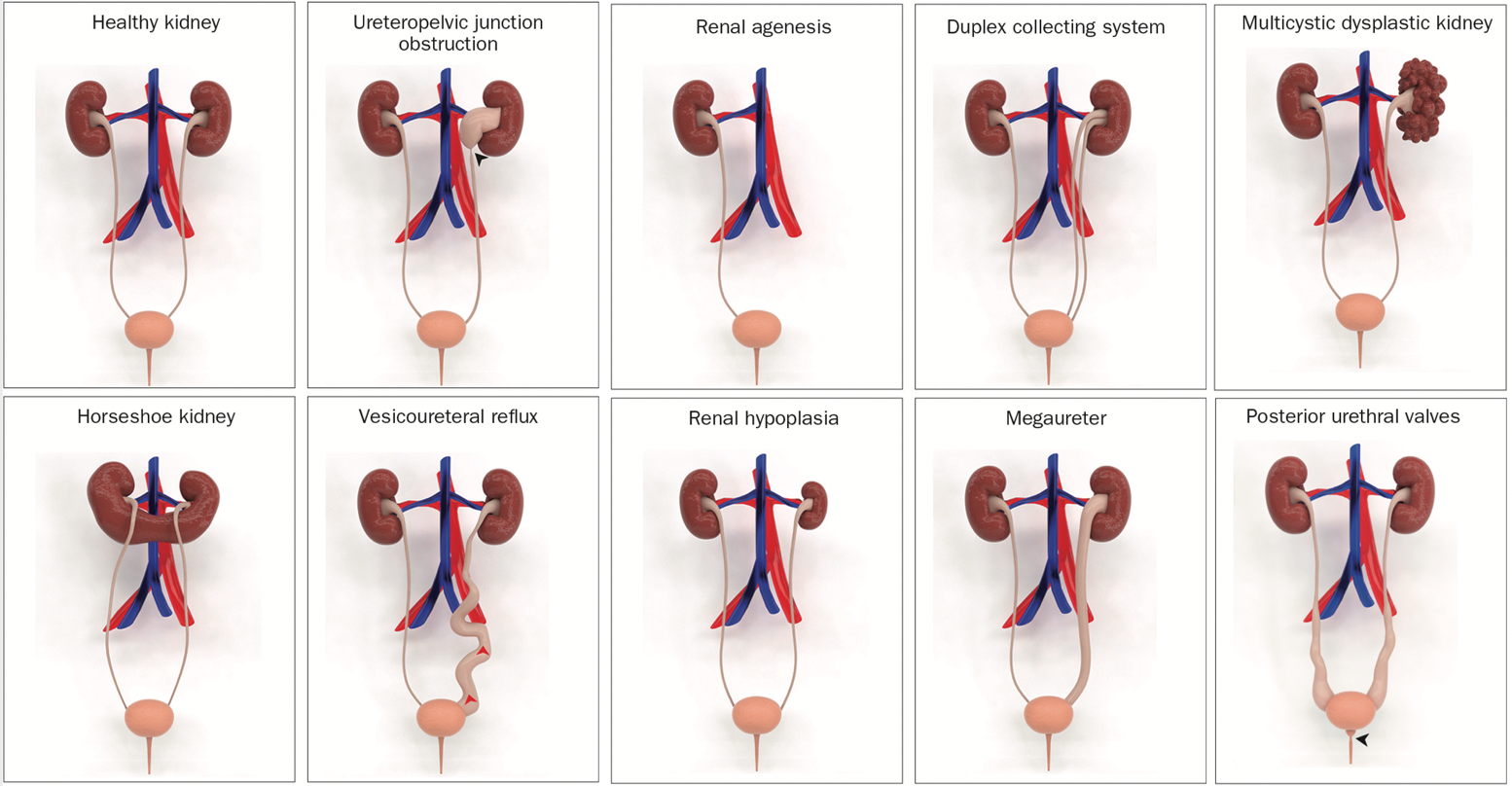
Figure 1 Modèles 3D illustrant les anomalies congénitales du rein et des voies urinaires (CAKUT).3
Embryologie
Le développement des reins et des voies urinaires chez le fœtus implique un processus complexe qui débute dès les premières semaines de la grossesse et comprend la formation et la différenciation de plusieurs structures. Ce processus commence par la formation des bourgeons urétéraux, qui émergent du mésoderme intermédiaire et donnent naissance aux tubes collecteurs et, en fin de compte, aux reins. Les reins se développent également à partir d’une structure appelée le métanéphros, qui se forme à partir du mésoderme chez l’embryon.
Au cours du développement, les bourgeons urétéraux et le métanéphros doivent s’unir correctement et se différencier pour former des reins fonctionnels et des voies urinaires. Des perturbations de ce processus peuvent entraîner une gamme d’anomalies congénitales.
Les CAKUT peuvent être classées selon la localisation (reins, uretères, vessie ou urètre), selon la cause (mutations génétiques, expositions environnementales, infections pendant la grossesse, etc.), selon le mode d’hérédité, et selon la gravité (Tableau 1). Dans 10 % à 25 % des cas, les CAKUT sont attribuées à des affections génétiques. Les CAKUT présentent des manifestations phénotypiques complexes en association avec des affections génétiques (Figure 2 et Figure 3).4 Il est possible que des individus présentant les mêmes anomalies du génome se manifestent par différentes formes de CAKUT, et que des anomalies phénotypiques similaires puissent résulter de différentes affections génétiques, et s’accompagner de maladies extrarénales.2
Tableau 1 Catégories des anomalies CAKUT
| Rein | Système collecteur |
|---|---|
| Nombre : agénésie rénale | Obstruction de la jonction pyélo-urétérale |
| Morphologie : hypoplasie rénale, dysplasie, RMCD | Reflux vésico-urétéral |
| Position : rein en fer à cheval, rein ectopique, rein pelvien | Méga-uretère |
| Duplicité du système collecteur | |
| VUP |
Plusieurs malformations du parenchyme rénal se traduisent par un défaut du développement normal des néphrons, notamment la dysplasie kystique, la dysplasie rénale, l’agénésie rénale, la dysgénésie tubulaire rénale et la dysgénésie tubulaire rénale.
Polykystose rénale
Le terme « polykystose rénale » est généralement utilisé pour décrire une maladie dans laquelle il existe de multiples kystes des reins sans dysplasie associée.
La polykystose rénale autosomique dominante (ADPKD) et la polykystose rénale autosomique récessive (ARPKD) sont les deux types les plus courants de polykystose rénale. Outre le mode de transmission, qu’il soit dominant ou récessif, ARPKD et ADPKD diffèrent par leurs manifestations cliniques et leur pronostic. L’ADPKD est la forme la plus fréquente de PKD, et elle est due à des mutations des gènes PKD1 ou PKD2.
Gènes impliqués dans le développement de la maladie kystique rénale:5,6
- PKD1: Ce gène est associé à l’ADPKD. Les mutations du gène PKD1 sont responsables d’environ 85 % des cas d’ADPKD.
- PKD2: Ce gène est également associé à l’ADPKD. Les mutations du gène PKD2 sont responsables d’environ 15 % des cas d’ADPKD.
- HNF1β: Les mutations de ce gène sont associées à une polykystose rénale autosomique dominante de type 2 (ADPKD2).
- REN: Ce gène est associé à une polykystose rénale infantile.
- MUC1: Les mutations de ce gène sont associées à une maladie rénale kystique médullaire liée à MUC1.
- INVS: Les mutations de ce gène sont associées à une maladie rénale kystique médullaire liée à la néphronophtise.
- NPHP1, NPHP3, NPHP4, NPHP5, NPHP6, NPHP7, NPHP8, NPHP9: Ces gènes sont associés à une maladie rénale kystique médullaire liée à la néphronophtise.
- BMP7: Les mutations de ce gène sont associées à une maladie rénale kystique médullaire liée à la néphronophtise
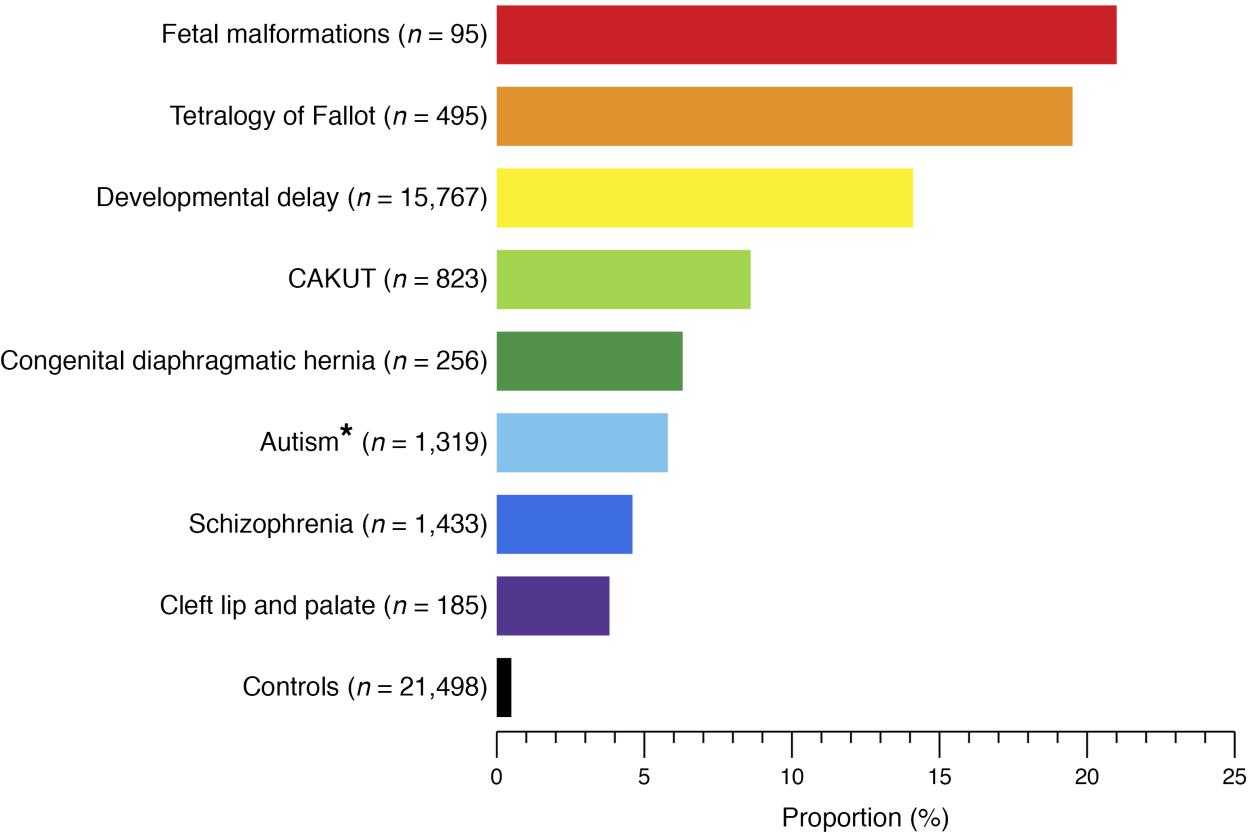
Figure 2 Proportion de patients présentant des syndromes génomiques connus dans différents phénotypes du développement humain et chez des témoins sains.4
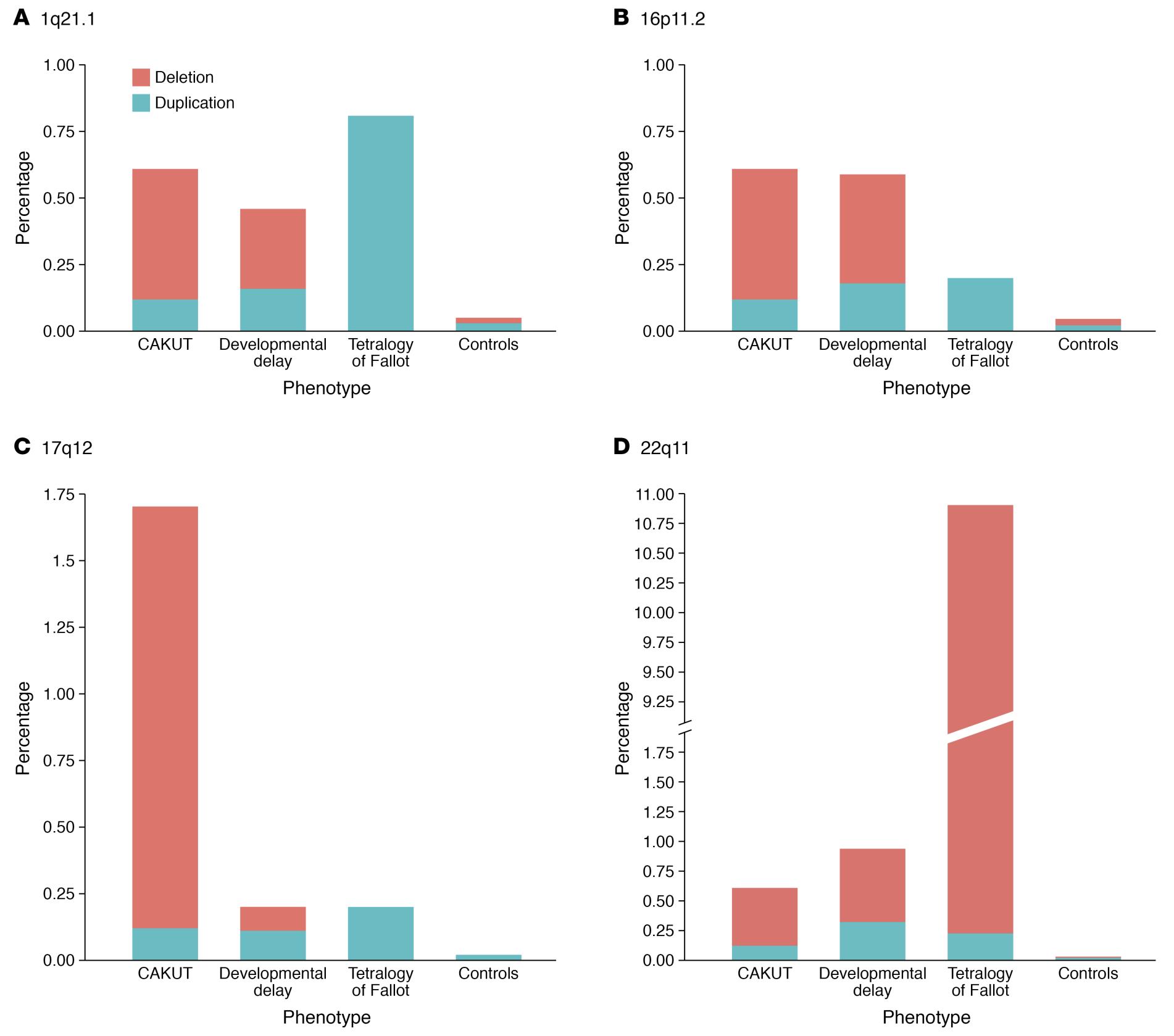
Figure 3 Différences et similitudes dans la prévalence des quatre loci CNV les plus fréquemment impliqués chez les patients atteints de CAKUT.4
MCDK
Le MCDK est une maladie kystique congénitale non héréditaire, un type de dysplasie rénale qui entraîne de multiples kystes non communicants séparés par un parenchyme dysplasique. La prévalence du MCDK est d’environ 1 sur 3 600 à 1 sur 4 300 naissances vivantes.7,8 La cause du MCDK est inconnue, mais on pense qu’elle pourrait être liée à une formation anormale du bourgeon urétéral, à une exposition à des tératogènes ou à une obstruction des voies urinaires. Le MCDK est souvent détecté par échographie prénatale. Comme il ne provoque souvent ni symptômes ni complications, il n’est généralement identifié après la naissance que s’il existe une masse perceptible ou s’il est découvert fortuitement lors d’un examen d’imagerie pour une autre affection.
Parmi les gènes associés à la MCDK figurent :9,10,11
- HNF1β: Ce gène code une protéine appelée facteur nucléaire hépatocytaire 1 bêta, qui joue un rôle dans le développement et la fonction du rein et d’autres organes. Des mutations de HNF1β peuvent entraîner un éventail d’anomalies rénales, y compris le MCDK.
- PAX2: Ce gène code une protéine appelée boîte appariée 2, qui est importante pour le développement de l’uretère, le tube qui transporte l’urine du rein à la vessie. Des mutations de PAX2 peuvent provoquer un MCDK et d’autres anomalies rénales.
- SIX1: Ce gène code une protéine appelée sine oculis homeobox 1, qui est impliquée dans le développement de divers organes, y compris le rein. Des mutations de SIX1 peuvent provoquer un MCDK et d’autres anomalies rénales.
- WT1: Ce gène code une protéine appelée tumeur de Wilms 1, qui est importante pour le développement du rein et d’autres organes. Des mutations de WT1 peuvent provoquer un MCDK et d’autres anomalies rénales.
Le MCDK est généralement pris en charge de façon conservatrice (c.-à-d., surveillance), car la plupart des cas ne s’accompagnent d’aucune complication à long terme. Les patients présentant des anomalies controlatérales peuvent développer une altération de la fonction rénale au fil du temps. Ainsi, un suivi à long terme est nécessaire.
Agénésie rénale
Comparativement à l’agénésie rénale bilatérale, l’agénésie rénale unilatérale survient dans une grossesse sur 1 000 et a un pronostic favorable. On estime qu’entre 0,1 % et 0,3 % des 1 000 naissances sont affectées par l’agénésie rénale bilatérale, laquelle est généralement associée à de faibles taux de survie. Plusieurs modes de transmission ont été décrits dans les familles présentant une agénésie rénale, qui peut survenir comme anomalie isolée ou s’intégrer dans un syndrome.12
Une agénésie rénale unilatérale peut être causée par des mutations dans plusieurs gènes, notamment ATRX, RET, BMP4, FRAS1, FREM1, GRIP1, GLI3, UPK3A, et d’autres. En outre, une agénésie rénale bilatérale peut être causée par des mutations des gènes RET, FGF20, ITGA8. Gènes apparentés répertoriés dans le Tableau 2.13,14
Dysplasie rénale
L’hypodysplasie rénale résulte d’un arrêt du développement du blastème rénal métanéphrogène au cours du premier trimestre de la vie fœtale. Cependant, l’étiologie de l’hypodysplasie rénale reste inconnue; des anomalies vasculaires in utero ainsi que des anomalies du développement d’origine génétique sont deux mécanismes possibles. L’hypodysplasie rénale peut constituer une découverte isolée ou faire partie d’un syndrome génétique associé à des malformations des voies urinaires. Une analyse des gènes du développement (HNF1β, PAX2, EYA1, SIX1 et SALL1) impliqués dans la dysplasie rénale, réalisée sur une large cohorte d’enfants atteints d’hypodysplasie rénale, a été effectuée dans le cadre de l’étude ESCAPE. Selon cette étude, des mutations de PAX2 ont été identifiées chez 15 % des patients présentant une pathologie rénale, et les patients porteurs de délétions génétiques ou de mutations de gènes de grande taille présentaient des phénotypes rénaux variables. De plus, 22 % de l’ensemble des patients atteints d’hypodysplasie rénale kystique présentaient une mutation de HNF1, ce qui suggère que les individus présentant une dysplasie rénale kystique devraient être dépistés pour des mutations de HNF1.15 À l’échographie rénale, une réduction de la taille rénale et une perte de la différenciation cortico-médullaire sont des signes cliniquement évocateurs d’hypodysplasie rénale. Une maladie rénale terminale peut se développer chez certains patients atteints d’hypodysplasie rénale au fil du temps. Il n’existe pas de traitement spécifique de l’hypodysplasie rénale. Il est important d’assurer un suivi et une prise en charge de la fonction rénale sur plusieurs années. Chez les patients qui évoluent vers une maladie rénale terminale, la transplantation rénale est l’option privilégiée.
Jonction pyélo-urétérale
La cause pathologique la plus fréquente d’hydronéphrose néonatale est l’obstruction de la portion proximale de l’uretère. L’incidence globale est de 1:1 500, avec un ratio de 2:1 chez les nouveau-nés de sexe masculin par rapport à ceux de sexe féminin.16 Une obstruction de la JPU est habituellement due à une sténose intrinsèque ou, plus rarement, à un vaisseau croisant entraînant une compression extrinsèque au niveau de la portion proximale de l’uretère. Un mécanisme possible est l’existence d’une perturbation embryologique de l’uretère proximal qui altère le développement de la musculature circulaire, en modifiant les fibres de collagène et leur composition entre et autour des cellules musculaires.
Plusieurs mutations monogéniques sont impliquées dans l’OJPU, notamment Id2, PAX2, EYA, AGTR2, BMP4, SOX17, CHD1L et DSTYK.17,18 Il a été rapporté que Id2 et Adamts1 produisent un phénotype semblable au phénotype d’OJPU humain. Les souris dépourvues d’Id2 présentent également une insertion urétérale haute dans le bassinet rénal.19
Reflux vésico-urétéral
Plusieurs gènes ont été identifiés comme étant associés au développement du RVU. Certains de ces gènes interviennent dans le développement et la fonction du muscle lisse urétéral. D’autres gènes sont impliqués dans le développement et la fonction de la vessie. La forte prévalence du RVU chez les jumeaux, les frères et sœurs et la descendance suggère une base génétique pour cette maladie. Il a été établi, sur la base des résultats d’études génétiques menées dans diverses populations, que le RVU est une maladie génétiquement hétérogène. Une étude de liaison et d’association à l’échelle du génome a rattaché le RVU primaire non syndromique à la région 10q26 du génome.20,21 D’autres gènes qui ont été associés au développement du RVU comprennent le gène FOXC1, le gène MYH11 et le gène ACTN4. Ces gènes jouent divers rôles dans le développement et la fonction des voies urinaires.22,23 Une étude d’association pangénomique utilisant la plus vaste analyse des variations du nombre de copies et l’étude d’association pangénomique du RVU aboutit à l’identification de cinq loci qui pourraient être associés au RVU, notamment WDPCP, OTX1, BMP5, WDPCP et WNT5, avec des effets importants. WNT5 pourrait jouer un rôle dans le développement du système urogénital. Une étude d’association pangénomique utilisant la plus vaste analyse des variations du nombre de copies et l’étude d’association pangénomique du RVU aboutit à l’identification de cinq loci qui pourraient être associés au RVU, notamment WDPCP, OTX1, BMP5, WDPCP et WNT5, avec des effets importants. WNT5 pourrait jouer un rôle dans le développement du système urogénital.24
Valves de l’urètre postérieur
Une valve urétrale postérieure est une anomalie congénitale des voies urinaires mettant en jeu le pronostic vital, généralement découverte au cours de la période néonatale et qui, malgré un traitement optimal, entraîne des taux élevés d’insuffisance rénale. On estime que l’incidence des VUP est de 1 pour 7 000–8 000 naissances vivantes.25
Il existe un large éventail de rapports indiquant que la PUV est la principale cause de maladie rénale chronique chez les nouveau-nés de sexe masculin, et l’ESRD peut survenir dans une proportion allant de 5 % à 64 % des cas.26,27 Une revue systématique a rapporté que la maladie rénale chronique (CKD) et la maladie rénale terminale (ESKD) sont associées chez les patients atteints de PUV à des risques allant jusqu’à 32 % et 20 %, respectivement.28
À ce jour, peu de gènes associés aux valves de l’urètre postérieur ont été identifiés. Le syndrome de Prune-Belly n’a été associé à HNF1B que dans 3 % des cas reconnus, et la plupart des personnes diagnostiquées n’ont pas de cause génétique. Parmi les anomalies CAKUT associées à la trisomie 21 figurent les valves de l’urètre postérieur, la pyélectasie et les méga-uretères.29,30
Tableau 2 Un résumé des gènes impliqués dans le développement des CAKUT.31,32,33,22,23,34,35,36
| Anomalie | Gènes |
|---|---|
| Hypoplasie/Dysplasie rénale | ACE, AGT, ATRX, CHD7, ESCO2, EYA1, SIX1, FRAS1, FREM2, GATA3, GLI3, GPC3, GRIP1, HNF1β, JAG1, KAL1, LRP4, MKS1–4, NIPBL, PAX2, PBX1, REN, SALL1, SIX5 |
| Agénésie rénale | ATRX, BMP4, CBP/EP300, CHD7, ESCO2, FGF20, FRAS1, FREM2, GATA3, GRIP1, GLI3, HNF1β, ITGA8, JAG1, KAL1, KAL2, RET, SALL1, VANGL1 |
| Dysplasie kystique | CBP/EP300, DHCR7, EVC, EVC2, FRAS1, FREM2, GPC-3, HNF1β, JAG1, KAL1, KAL2, MKS1–4, NS1, PEX, PAX2, WT1 |
| Système dupliqué | NS1 |
| Rein en fer à cheval/Rein ectopique | GLI3, NIPBL, VANGL1 |
| Hydronéphrose, JPU | ACE, Adamts-1, AGT, ATRX, BMP4, CHD1L, CHD7, DHCR7, DSTYK, ESCO2, EYA, FRAS1, FREM2, GLI3, HSPG2, JAG1, Id2, NS1, PAX2, PEX, RET, SOX17, VANGL1 |
| Hydro-uretère, Méga-uretère | CHD7, GLI3 |
| RVU | ATRX, DHCR7, EYA1, FOX1, GATA3, HOXA13, HPSE2, JAG1, KAL1, KAL2, MYH11, NIPBL, PAX2, SALL1, SIX1, SIX5 |
| VUP | BNC2, SALL1 |
Importance clinique et implications futures des connaissances sur les gènes
À mesure que le domaine de l’évaluation génétique continue de progresser, il est probable que nous verrons une capacité accrue à identifier les causes génétiques des CAKUT et à prédire la probabilité de développer ces affections. Cela pourrait conduire à un diagnostic et un traitement plus précoces, ce qui pourrait améliorer le pronostic des personnes atteintes.
Il convient de noter que les tests génétiques ne sont pas toujours nécessaires pour établir le diagnostic de CAKUT. Un diagnostic définitif est généralement posé sur la base d’une combinaison de la présentation clinique, des antécédents médicaux et des examens d’imagerie. Les tests génétiques pour la CAKUT sont habituellement réalisés dans un centre spécialisé sous la supervision d’un généticien clinicien, car l’interprétation de ces résultats peut être complexe et avoir des implications pour les décisions reproductives. Plusieurs laboratoires (PreventionGenetics, Invitae, Blueprint Genetics, etc.) proposent des tests génétiques pour différentes affections de la CAKUT. Leur panel de tests pour la CAKUT comprend des analyses génétiques de multiples gènes associés aux troubles CAKUT, notamment HNF1B, NPHP1 et UPK1B. Les tests génétiques pour ces affections peuvent aider à confirmer un diagnostic, fournir des informations sur la gravité de l’affection et contribuer au conseil génétique et à la planification familiale. Il convient de noter que d’autres entreprises et laboratoires de tests génétiques peuvent également proposer un panel CAKUT. Étant donné que la CAKUT est un groupe d’affections causées par différentes mutations génétiques, le test de panel génétique pour la CAKUT proposé par certaines entreprises peut ne pas inclure toutes les causes génétiques de la CAKUT, et différents laboratoires peuvent avoir une liste de gènes couverts différente.
Une compréhension complète des composantes génétiques du CAKUT est essentielle pour élaborer des stratégies précises de tests génétiques. Par ailleurs, cette compréhension contribuera à guider la prise de décision clinique en matière de tests génétiques. En incluant des résultats cliniques importants et la fonction rénale, des études longitudinales sont nécessaires pour examiner les effets à long terme du diagnostic précoce et de la prise en charge du CAKUT.
Conclusion et résumé
Les anomalies congénitales du rein et des voies urinaires (CAKUT) sont des anomalies structurelles qui affectent les reins et les voies urinaires. On estime qu’il y a environ 4–60 cas pour 10 000 naissances. Le diagnostic de CAKUT est généralement posé lors d’une échographie prénatale ou après la naissance chez les nouveau-nés présentant des signes et symptômes cliniques associés. L’anomalie la plus fréquente est l’obstruction de la jonction pyélo-urétérale, qui touche environ 20 % des personnes atteintes de cette affection. Le développement des reins et des voies urinaires implique la formation et la différenciation de plusieurs structures, notamment les bourgeons urétéraux et le métanéphros, qui doivent se connecter correctement pour former des reins et des voies urinaires fonctionnels. Des perturbations de ce processus peuvent conduire à des anomalies CAKUT. Il existe diverses catégories de troubles CAKUT, notamment ceux affectant le nombre, la morphologie et la position des reins, et ceux affectant le système collecteur. Dans 10–25 % des cas, les CAKUT sont dus à des troubles génétiques. Plusieurs gènes associés au développement de diverses maladies rénales ont été identifiés. Des recherches sont en cours pour améliorer notre compréhension des causes des CAKUT et pour élaborer de meilleures stratégies de prévention et de traitement. Malgré ses limites et difficultés actuelles, le diagnostic génétique moléculaire pourrait offrir l’espoir d’améliorer à l’avenir la prise en charge clinique des patients atteints de CAKUT.
Points clés
- Les anomalies congénitales du rein et des voies urinaires (CAKUT) sont un groupe d’anomalies structurelles qui affectent les reins et les voies urinaires, avec une estimation de 4 à 60 cas pour 10 000 naissances dans la population générale.
- Le développement des reins et des voies urinaires chez le fœtus implique un processus complexe qui débute dès les premières semaines de la grossesse et comprend la formation et la différenciation de plusieurs structures. Des perturbations de ce processus peuvent entraîner un éventail d’anomalies congénitales.
- La prévalence des CAKUT chez les prématurés était de 2 % dans une large cohorte récemment rapportée comprenant 409 704 nourrissons.
- L’anomalie la plus fréquente est l’obstruction de la jonction pyélo-urétérale, qui touche environ 20 % des personnes atteintes.
- Dans 10 à 25 % des cas, les CAKUT sont attribuées à des affections génétiques.
- L’évaluation génétique progresse, augmentant la capacité à identifier les causes génétiques des CAKUT et à prédire la probabilité de développer ces affections.
- Les tests génétiques ne sont pas toujours nécessaires pour diagnostiquer les CAKUT ; le diagnostic repose généralement sur une combinaison de la présentation clinique, des antécédents médicaux et des examens d’imagerie.
- Une compréhension complète des composantes génétiques des CAKUT est essentielle pour élaborer des stratégies de tests génétiques précises et guider la prise de décision clinique en matière de tests génétiques.
- Des études longitudinales sont nécessaires pour examiner les effets à long terme du diagnostic précoce et de la prise en charge des CAKUT.
Références
- Murugapoopathy V, Gupta IR. A Primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin J Am Soc Nephrol 2020; 15 (5): 723–731. DOI: 10.2215/cjn.12581019.
- Hays T, Thompson MV, Bateman DA, Sahni R, Tolia VN, Clark RH, et al.. The Prevalence and Clinical Significance of Congenital Anomalies of the Kidney and Urinary Tract in Preterm Infants. JAMA Netw Open 2022; 5 (9): e2231626. DOI: 10.1001/jamanetworkopen.2022.31626.
- Nicolaou N, Renkema KY, Bongers EMHF, Giles RH, Knoers NVAM. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol 2015; 11 (12): 720–731. DOI: 10.1038/nrneph.2015.140.
- Srivastava S, Molinari E, Raman S, Sayer JA. Many Genes–One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front Pediatr 2018; 5. DOI: 10.3389/fped.2017.00287.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. 8 (1): 25–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 2018; 128 (1): 4–15. DOI: 10.1172/jci95300.
- Gordon AC, Thomas DF, Arthur RJ, Irving HC. Multicystic dysplastic kidney: Is nephrectomy still appropriate? J Pediatr Surg 1988; 24 (9): 951. DOI: 10.1016/s0022-3468(89)80673-6.
- Schreuder MF, Westland R, Wijk JAE van. Unilateral multicystic dysplastic kidney: a meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol Dial Transplant 2009; 24 (6): 1810–1818. DOI: 10.1093/ndt/gfn777.
- Kopač M, Kordič R. Associated Anomalies and Complications of Multicystic Dysplastic Kidney. Pediatr Rep 2022; 14 (3): 375–379. DOI: 10.3390/pediatric14030044.
- Fletcher J, Hu M, Berman Y, Collins F, Grigg J, McIver M, et al.. Multicystic Dysplastic Kidney and Variable Phenotype in a Family with a Novel Deletion Mutation ofPAX2. J Am Soc Nephrol 2005; 16 (9): 2754–2761. DOI: 10.1681/asn.2005030239.
- Chang Y-M, Chen C-C, Lee N-C, Sung J-M, Chou Y-Y, Chiou Y-Y. PAX2 Mutation-Related Renal Hypodysplasia: Review of the Literature and Three Case Reports. Front Pediatr 2022; 9 (765929). DOI: 10.3389/fped.2021.765929.
- Bienstock JL, Birsner ML, Coleman F, Hueppchen NA. Successful In Utero Intervention for Bilateral Renal Agenesis. Obstet Gynecol 2014; 124 (2): 413–415. DOI: 10.1097/aog.0000000000000339.
- Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, et al.. Faculty Opinions recommendation of Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2012; 1 (2): 96–200. DOI: 10.3410/f.13934957.15391059.
- Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal Aplasia in Humans Is Associated with RET Mutations. Am J Hum Genet 2008; 82 (2): 344–351. DOI: 10.1016/j.ajhg.2007.10.008.
- Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al.. Faculty Opinions recommendation of Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2006; 7 (10): 864–870. DOI: 10.3410/f.1047767.508721.
- LEBOWITZ ROBERTL, GRISCOM NTHORNE. Neonatal Hydronephrosis. Radiol Clin North Am 1977; 15 (1): 49–59. DOI: 10.1016/s0033-8389(22)02539-8.
- AC SJ, DM M, oes S, AC S. Congenital Anomalies of the Kidney and Urinary Tract: An Overview. Congenital Anomalies of the Kidney and Urinary Tract 2014; 02 (4): 1–13. DOI: 10.1007/978-3-319-29219-9_1.
- Avanoglu A, Tiryaki S. Embryology and Morphological (Mal)Development of UPJ. Front Pediatr 2020; 8: 137. DOI: 10.3389/fped.2020.00137.
- Aoki Y, Mori S, Kitajima K, Yokoyama O, Kanamaru H, Okada K, et al.. Id2 haploinsufficiency in mice leads to congenital hydronephrosis resembling that in humans. Genes Cells 2004; 9 (12): 1287–1296. DOI: 10.1111/j.1365-2443.2004.00805.x.
- Williams G, Fletcher JT, Alexander SI, Craig JC. Vesicoureteral reflux. Journal of the American Society of Nephrology 2008; 19 (5): 847–862. DOI: 10.1681/asn.2007020245.
- Darlow JM, Darlay R, Dobson MG, Stewart A, Charoen P, Southgate J, et al.. Genome-wide linkage and association study implicates the 10q26 region as a major genetic contributor to primary nonsyndromic vesicoureteric reflux. Sci Rep 2017; 7 (1): 59. DOI: 10.1038/s41598-017-15062-9.
- Woolf AS, Lopes FM, Ranjzad P, Roberts NA. Congenital Disorders of the Human Urinary Tract: Recent Insights From Genetic and Molecular Studies. Front Pediatr 2019; 7 (136). DOI: 10.3389/fped.2019.00136.
- Wu C-HW, Mann N, Nakayama M, Connaughton DM, Dai R, Kolvenbach CM, et al.. Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT). Genet Med 2020; 22 (10): 1673–1681. DOI: 10.1038/s41436-020-0844-z.
- Verbitsky M, Krithivasan P, Batourina E, Khan A, Graham SE, Marasà M, et al.. Review of: "Genome-wide association study followed by trans-ancestry meta-analysis identify 17 new risk loci for schizophrenia". J Am Soc Nephrol 2021; 32 (4): 805–820. DOI: 10.32388/jqb9pr.
- Thakkar D, Deshpande AV, Kennedy SE. Epidemiology and demography of recently diagnosed cases of posterior urethral valves. Pediatr Res 2014; 76 (6): 560–563. DOI: 10.1038/pr.2014.134.
- Warshaw BL, Edelbrock HH, Ettenger RB, Malekzadeh MH, Pennisi AJ, Uittenbogaart CH, et al.. Renal transplantation in children with obstructive uropathy. J Pediatr Surg 1980; 15 (6): 986. DOI: 10.1016/s0022-3468(80)80355-1.
- Roth KS, Carter WH, Chan JCM. Obstructive Nephropathy in Children: Long-Term Progression After Relief of Posterior Urethral Valve. Pediatrics 2001; 107 (5): 1004–1010. DOI: 10.1542/peds.107.5.1004.
- Hennus PML, Kort LMO de, Bosch JLH, Jong TPVM de, Heijden GJMG van der. A Systematic Review on the Accuracy of Diagnostic Procedures for Infravesical Obstruction in Boys. PLoS One 2014; 9 (2): e85474. DOI: 10.1371/journal.pone.0085474.
- Rodriguez MM. Congenital anomalies of the kidney and urinary tract (CAKUT). Lijec Vjesn 2014; 144 (Supp 1). DOI: 10.26800/lv-144-supl1-26.
- Postolache L, Parsa A, Simoni P, Boitsios G, Ismaili K, Schurmans T, et al.. Widespread kidney anomalies in children with Down syndrome. Pediatr Nephrol 2022; 37 (10): 2361–2368. DOI: 10.1007/s00467-022-05455-y.
- Uy N, Reidy K. Developmental Genetics and Congenital Anomalies of the Kidney and Urinary Tract. J Pediatr Genet 2016; 05 (01): 051–060. DOI: 10.1055/s-0035-1558423.
- Kolvenbach CM, Dworschak GC, Frese S, Japp AS, Schuster P, Wenzlitschke N, et al.. Expanding congenital abnormalities of the kidney and urinary tract (CAKUT) genetics: basonuclin 2 (BNC2) and lower urinary tract obstruction. Ann Transl Med 2019; 7 (S6): S226–s226. DOI: 10.21037/atm.2019.08.73.
- Heidet L, Morinière V, Henry C, De Tomasi L, Reilly ML, Humbert C, et al.. Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 2017; 28 (10): 2901–2914. DOI: 10.1681/asn.2017010043.
- Talati AN, Webster CM, Vora NL. Prenatal genetic considerations of congenital anomalies of the kidney and urinary tract (CAKUT). Prenat Diagn 2019; 39 (9): 679–692. DOI: 10.1002/pd.5536.
- Puri P, Gosemann J-H, Darlow J, Barton DE. Genetics of vesicoureteral reflux. Nat Rev Urol 2011; 8 (10): 539–552. DOI: 10.1038/nrurol.2011.113.
- Chen F. Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol 2009; 24 (9): 1621–1632. DOI: 10.1007/s00467-008-1072-y.
Dernière mise à jour: 2025-09-22 07:59