
40: DSD—Comprensión actual, estudio diagnóstico y tratamiento
Este capítulo durará aproximadamente 24 minutos para leer.
Introducción y Terminología
Los trastornos/diferencias del desarrollo sexual (DSD, también denominado intersexualidad) son afecciones congénitas en las que el sexo cromosómico, gonadal o fenotípico difiere de lo que se considera típicamente masculino o femenino. En 2006 se introdujo una nueva y amplia clasificación de los DSD mediante el “Consensus Statement on Management of Intersex Disorders.”1 Esta clasificación más reciente incorpora una amplia gama de afecciones, incluidas la hiperplasia suprarrenal congénita (CAH), el DSD ovotesticular y el síndrome de insensibilidad a los andrógenos (AIS). La definición de consenso de 2006 es más amplia que lo que anteriormente se denominaba afecciones intersexuales, e incluye también anomalías anatómicas como la extrofia cloacal y vesical y la agenesia vaginal, así como anomalías cromosómicas que no causan genitales atípicos (p. ej., síndrome de Klinefelter [47, XXY]). Tabla 1 ofrece una visión general de la nomenclatura previa y de las actualizaciones de terminología recomendadas propuestas en la declaración de consenso de 2006. Dada la amplia gama de afecciones bajo el ‘paraguas’ de los DSD, es fundamental un enfoque de atención individualizado y multidisciplinario.
Tabla 1 Nomenclatura de DSD revisada. Fuente: Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
| Anterior | Propuesto |
|---|---|
| Intersexo | DSD |
| Pseudohermafrodita masculino | 46,XY DSD |
| Pseudohermafrodita femenino | 46,XX DSD |
| Hermafrodita verdadero | DSD ovotesticular |
| Disgenesia gonadal mixta | Disgenesia gonadal mixta (sin cambios) |
| Varón XX o reversión sexual XX | 46,XX DSD testicular |
| Reversión sexual XY | 46,XY disgenesia gonadal completa |
Ha habido una adopción casi universal de la nueva terminología DSD por parte de la comunidad médica desde que fue introducida. Entre las personas afectadas por afecciones DSD, existe variabilidad en las preferencias terminológicas y una falta de consenso sobre exactamente qué afecciones deberían considerarse un DSD.2,3,4 En particular, algunos miembros de la comunidad con CAH se identifican como portadores de un trastorno endocrino, no de una condición DSD/intersexo.5 Tampoco está claro qué personas con hipospadias proximal deberían considerarse que tienen “un DSD”.6 Los clínicos que atienden afecciones DSD deben estar al tanto de la evolución y las controversias de la nomenclatura, y utilizar los términos que sus pacientes prefieran durante los encuentros médicos individuales. Para los fines de este capítulo, se utilizará la terminología actual, aceptada médicamente.
Embriología
Desarrollo típico de los genitales internos/externos
Todos los fetos en desarrollo comienzan de la misma manera. Las estructuras anatómicas para el desarrollo masculino o femenino y sus variaciones están presentes en las primeras semanas de la gestación (Tabla 2) incluyendo la cresta gonadal, los conductos de Wolff (mesonéfricos) y de Müller (paramesonéfricos), la cloaca y, posteriormente, el seno urogenital, el tubérculo genital y las prominencias labioescrotales. Figura 1 muestra los genes responsables del desarrollo de la gónada indiferenciada hacia un testículo o un ovario.
Tabla 2 Precursores embriológicos y estructuras típicas masculinas/femeninas.
| Estructura embriológica | Estructura típica femenina | Estructura típica masculina |
|---|---|---|
| Cresta gonadal | Ovario | Testículo |
| Conducto de Wolff (mesonéfrico) | Paroóforo, epoóforo, quiste del conducto de Gartner | Conducto deferente, epidídimo, vesículas seminales |
| Conducto de Müller (paramesonéfrico) | Trompas de Falopio, útero, vagina proximal | Apéndice testicular, utrículo prostático |
| Cloaca y seno urogenital subsiguiente | Vejiga, vagina distal, uretra | Vejiga, próstata, uretra |
| Tubérculo genital | Clítoris | Pene |
| Prominencias labioescrotales | Complejo labial | Escroto |
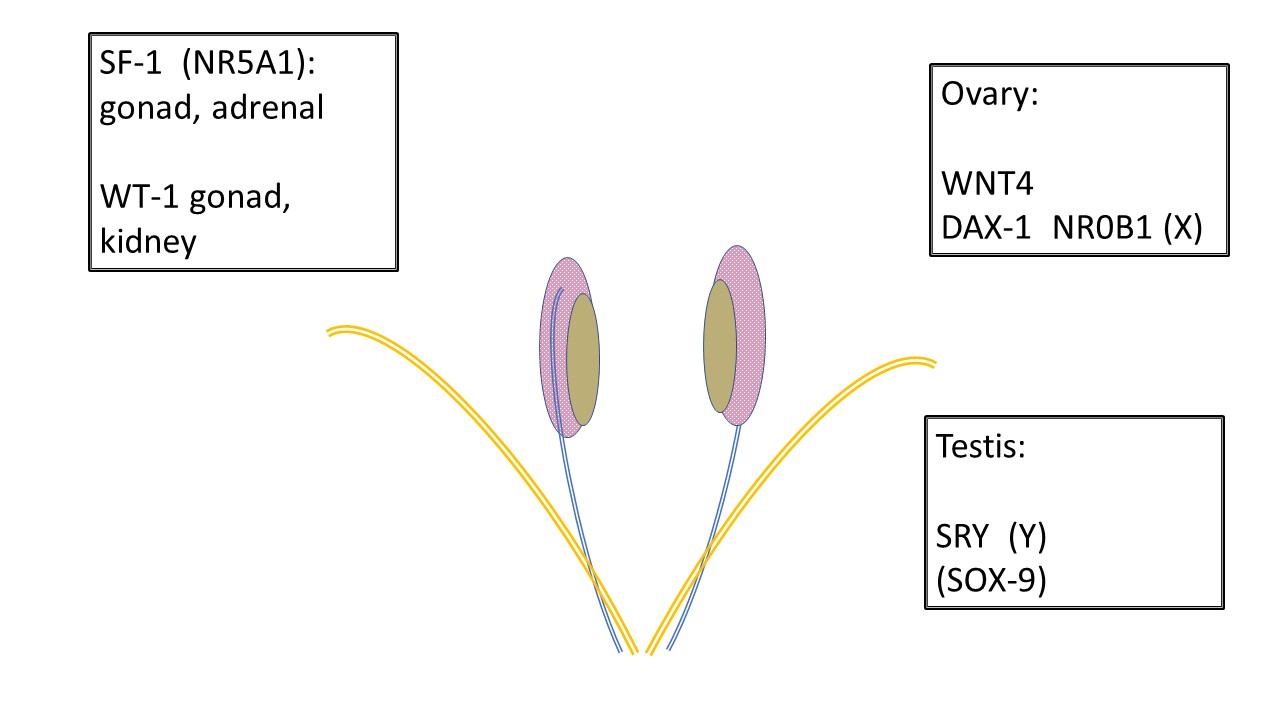
Figura 1 Desarrollo de las gónadas indiferenciadas. Todas las gónadas comienzan de la misma manera. Las células germinales primordiales migran a la cresta gonadal antes de las 6 semanas y la infraestructura que sustenta el desarrollo gonadal está además influida por diversos genes. SF-1 y WT-1 influyen en el desarrollo gonadal y en la posterior comunicación endocrina con los conductos de Wolff (mesonéfrico-azul) y de Müller (paramesonéfrico-naranja). Observe la relación entre el mesonefros (rosa), el conducto mesonéfrico y la gónada indiferenciada (beige). WNT4 es el gen determinante del ovario. DAX-1 se considera el “gen anti-testículo”. La duplicación de DAX-1 produce reversión sexual XY. SRY en el cromosoma Y es el gen determinante del testículo. SOX-9 favorece el desarrollo de las células de Sertoli, pero también tiene homología para participar en el desarrollo testicular en ausencia de SRY.
El desarrollo masculino típico (Figura 2) se inicia muy temprano en la gestación por el cromosoma Y, específicamente SRY, determinando el desarrollo testicular. WT-1, SF-1 están implicados en el desarrollo del testículo y del ovario, y SOX-9 está implicado en la diferenciación de las células de Sertoli. Las células de Leydig del testículo posteriormente comienzan la producción de testosterona aproximadamente a las 8 semanas de gestación y las células de Sertoli producen hormona antimülleriana (AMH), inhibiendo la maduración de los conductos de Müller en útero, trompas de Falopio y vagina superior. La testosterona dirige el crecimiento temprano del tubérculo genital y el desarrollo del conducto de Wolff en conducto deferente, epidídimo y vesícula seminal. El desarrollo o su ausencia en los sistemas de Wolff y de Müller ocurre como una acción paracrina más que sistémica, lo que explica el desarrollo asimétrico de las estructuras ductales cuando la composición gonadal es asimétrica. La conversión de testosterona a la más potente dihidrotestosterona (DHT) por la 5α-reductasa-2 ocurre en varios tejidos y es responsable de la maduración adicional del pene, la tubularización del seno urogenital y de la placa uretral, y el cierre del escroto hacia las 14 semanas de gestación. El crecimiento posterior de la estructura fálica ocurre debido a la disminución de los andrógenos fetales y al crecimiento somático.
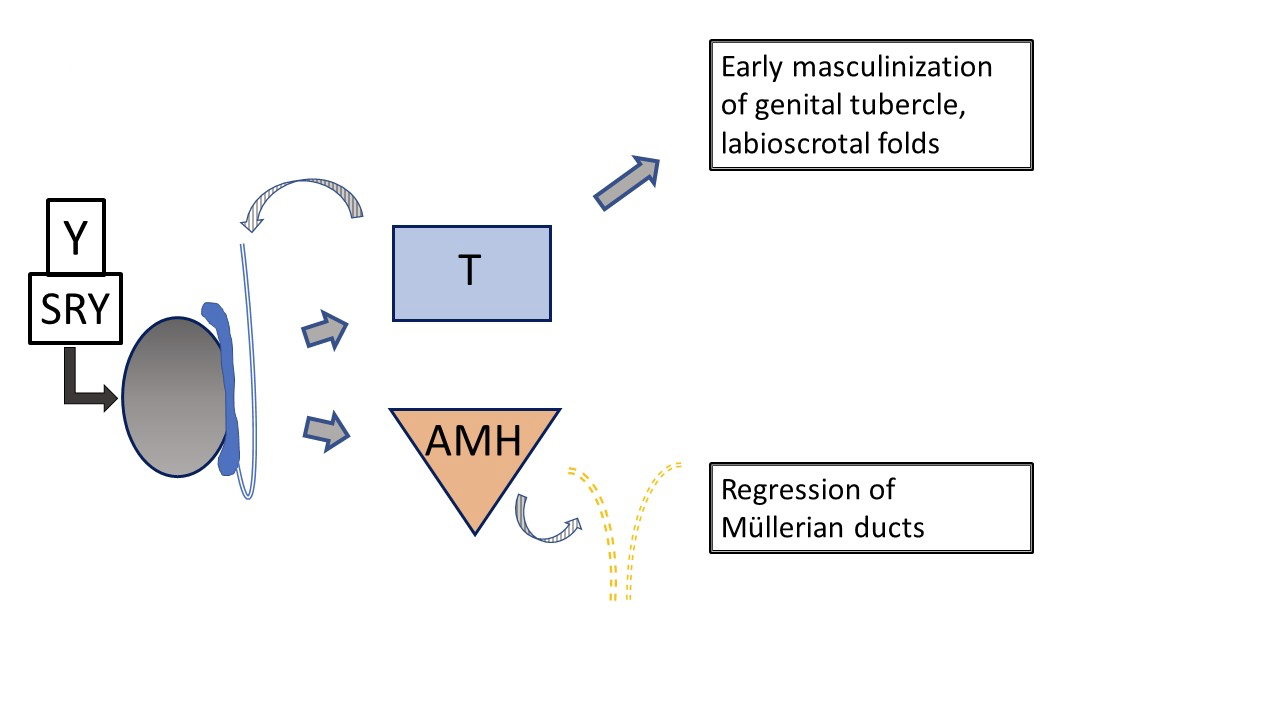
Figura 2 Desarrollo temprano típico del tracto genital masculino externo e interno, influido por el testículo fetal. La producción de testosterona y AMH por el testículo fetal da como resultado la masculinización temprana del tubérculo genital y la regresión de los conductos de Müller. El desarrollo posterior de los genitales externos y la uretra está influido por la conversión de testosterona a dihidrotestosterona. (no se muestra aquí, véase Figura 4).
El desarrollo femenino típico (Figura 3) a menudo se considera que ocurre por defecto dado que no está mediado por hormonas producidas por los testículos. La determinación ovárica no desempeña un papel destacado en el desarrollo embrionario de los genitales. Niveles bajos de andrógenos y AMH permiten el desarrollo femenino típico del tubérculo genital como clítoris, de los pliegues labioscrotales como complejo labial y del seno urogenital distal como uretra y vagina. Los cuerpos müllerianos pares migran y se fusionan para formar el útero. Juntos, estimulan el bulbo sinovaginal en el seno urogenital para iniciar el desarrollo de la porción inferior de la vagina.
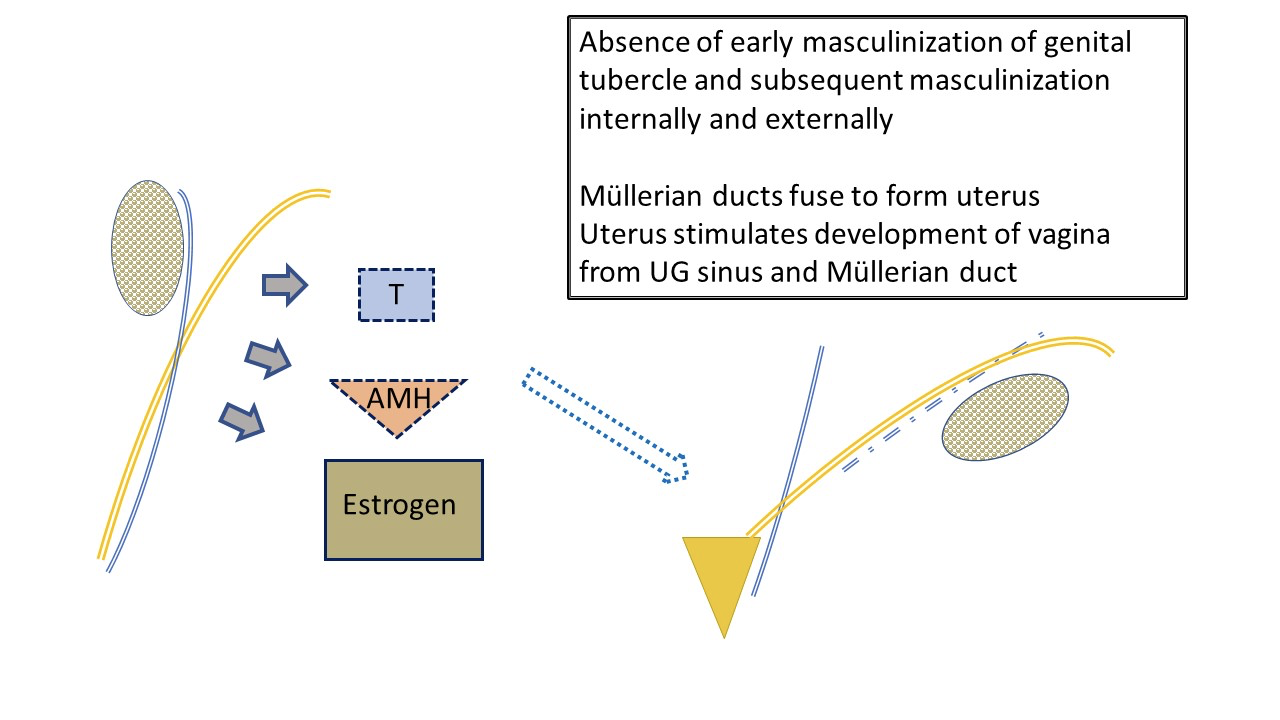
Figura 3 Desarrollo temprano típico del tracto genital femenino externo e interno, influido por el ovario fetal. Los niveles bajos de AMH y andrógenos producidos por el ovario dan lugar al desarrollo de la anatomía femenina típica: útero y trompas de Falopio, vagina superior. La producción fetal de estrógenos tiene menos impacto en el desarrollo de la vía femenina que un entorno con niveles mínimos de andrógenos y AMH. Los labios se verán afectados por los estrógenos maternos. Obsérvese también la relación definitiva de “agua bajo el puente” entre el uréter (derivado del conducto mesonéfrico) y la trompa de Falopio. Obsérvense restos del conducto mesonéfrico asociados con la trompa de Falopio.
Diferentes vías que pueden causar DSD
Las diferencias del desarrollo pueden ocurrir mediante atipicidad en:
- Determinación genética de las gónadas (p. ej., disgenesia gonadal mixta, DSD ovotesticular)
- Producción hormonal (p. ej., deficiencia de 5α-reductasa, deficiencia de 3-β-hidroxiesteroide deshidrogenasa, CAH por deficiencia de 21-hidroxilasa, o mutaciones del receptor de la hormona luteinizante [LH])
- Acción hormonal (p. ej., AIS completa o parcial)
- Variaciones en el desarrollo del tejido precursor/separación de la cloaca (p. ej., síndrome de Mayer-Rokitansky-Küster-Hauser [MRKH], extrofia cloacal, anomalías cloacales)
Incluso con un cariotipo 46,XX o 46,XY, no hay certeza de que el desarrollo siga la trayectoria típica femenina o masculina. En 46,XY por sí solo, se presentan numerosas diferencias debido a mutaciones del cromosoma Y que alteran la determinación o la función testicular, mutaciones somáticas que llevan a disgenesia del testículo en desarrollo, variaciones en la biosíntesis o conversión de testosterona y mutaciones ligadas al cromosoma X que afectan la función del receptor de andrógenos. El fenotipo externo, el desarrollo interno, el potencial de pubertad espontánea o de fertilidad y la identidad a lo largo del espectro de género pueden verse afectados. Al considerar las características de las diversas condiciones DSD descritas a continuación, es importante señalar que la producción y la acción hormonales se dan en un espectro. Por lo tanto, las deficiencias en la función del receptor o en la producción de hormonas y de enzimas convertidoras no son necesariamente déficits completos. El fenotipo puede variar ampliamente, incluso entre individuos con el mismo diagnóstico.
Fisiopatología
Las variaciones en el desarrollo de los genitales internos y externos están determinadas por mutaciones de los cromosomas sexuales y somáticas, la función gonadal resultante y la respuesta tisular. Cuando la función del receptor o la presencia de una enzima pertinente es atípica debido a una mutación genética conocida o aún no identificada, el efecto sobre los órganos diana puede ser completo (sin función evidente) o parcial. El fenotipo varía en consecuencia. Como ejemplo, diversos fenotipos de insensibilidad a los andrógenos (completa CAIS, parcial PAIS y leve MAIS: espectro del fenotipo genital desde femenino típico—ambiguo—masculino típico) se deben a numerosas mutaciones distintas del gen del receptor de andrógenos en el cromosoma X (trastorno recesivo ligado al X). Tabla 3 muestra un sistema de clasificación de DSD, dividido por cariotipo.
Tabla 3 Un ejemplo de clasificación de DSD
| TDS de los cromosomas sexuales | TDS 46,XY | TDS 46,XX |
|---|---|---|
| 45,X (síndrome de Turner y variantes) | Trastornos del desarrollo gonadal (testicular): (1) disgenesia gonadal completa (síndrome de Swyer); (2) disgenesia gonadal parcial; (3) regresión gonadal; y (4) TDS ovotesticular |
Trastornos del desarrollo gonadal (ovárico): (1) TDS ovotesticular; (2) TDS testicular (p. ej., SRY+, duplicación de SOX9); y (3) disgenesia gonadal |
| 47,XXY (síndrome de Klinefelter y variantes) | Trastornos en la síntesis o acción de andrógenos: (1) defecto de la biosíntesis de andrógenos (p. ej., deficiencia de 17-hidroxiesteroide deshidrogenasa, deficiencia de 5RD2, mutaciones de StAR); (2) defecto en la acción de andrógenos (p. ej., CAIS, PAIS); (3) defectos del receptor de la hormona luteinizante (p. ej., hipoplasia de células de Leydig, aplasia); y (4)trastornos de la hormona antimülleriana y del receptor de la hormona antimülleriana (síndrome de persistencia de los conductos de Müller) |
Exceso de andrógenos: (1) fetal (p. ej., deficiencia de 21-hidroxilasa, deficiencia de 11-hidroxilasa); (2) fetoplacentario (deficiencia de aromatasa, POR [oxidoreductasa P450]); y (3) materno (luteoma, exógeno, etc) |
| 45,X/46,XY (MGD, TDS ovotesticular) | Otros (p. ej., extrofia cloacal, atresia vaginal, MURCS [anomalías müllerianas, renales y de los somitas cervicotorácicos], otros síndromes) | |
| 46,XX/46,XY (quimérico, TDS ovotesticular) |
46,XX DSD
La forma más común de DSD 46,XX es la CAH, en la que la función enzimática suprarrenal está alterada. Figura 4 muestra la cascada típica de esteroides suprarrenales, que comienza con el colesterol. Los productos finales son la aldosterona, el cortisol y los esteroides sexuales. La rama de esteroides sexuales de la cascada termina con la androstenediona, que se convierte en una pequeña cantidad de testosterona. La producción de testosterona en las células de Leydig del testículo también comienza con el colesterol. En la CAH, la disminución de la función enzimática causa una reducción de los mineralocorticoides (aldosterona) y los glucocorticoides (cortisol), y una desviación hacia la producción de esteroides sexuales. El resultado es la masculinización de un feto con cariotipo 46,XX. Aunque es más fácil recordar una ausencia completa de la función enzimática y de los productos esteroideos suprarrenales derivados, la función puede estar solo parcialmente alterada y la retroalimentación en el eje hipotálamo-hipofisario puede verse afectada en consecuencia.
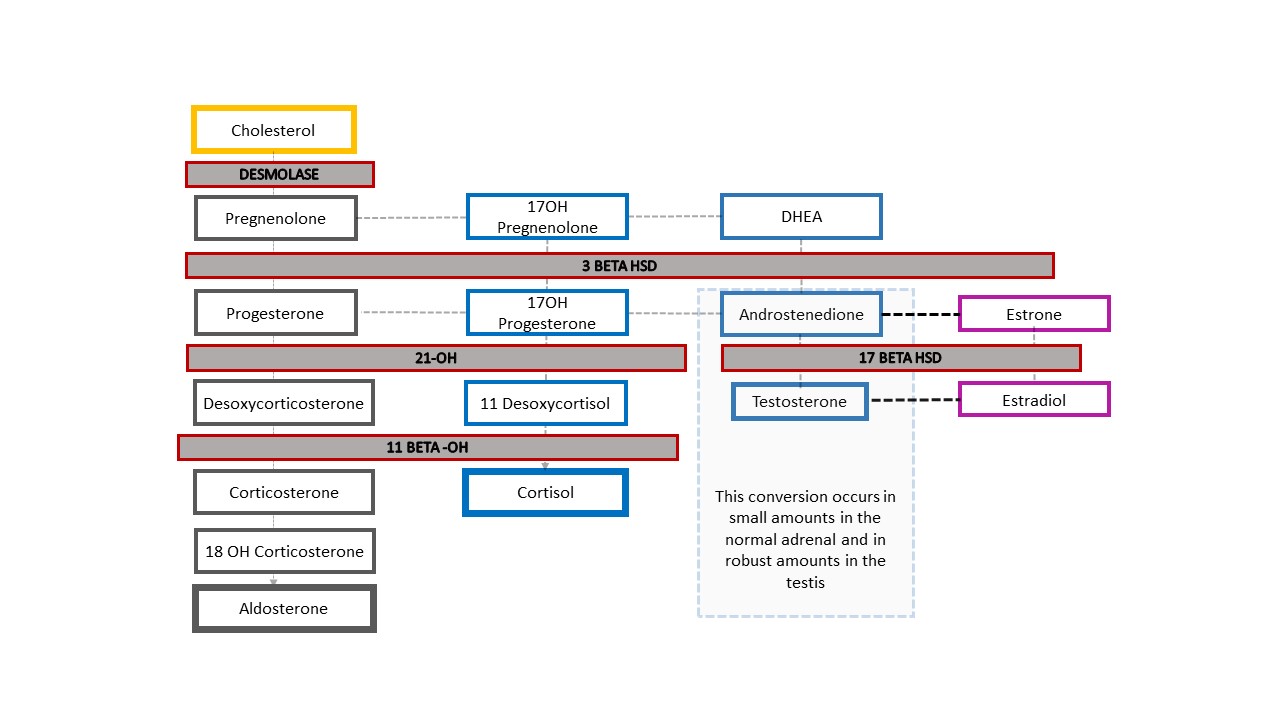
Figura 4 Cascada esteroidea y espectro de diagnósticos de HSC. Cualquier grado de defecto en la producción de una enzima provoca una disminución del producto posterior en la vía y una desviación de la vía hacia los esteroides sexuales. El eje hipotálamo-hipófiso-suprarrenal se estimula para compensar. El aumento de ACTH produce hiperplasia de la corteza suprarrenal y una producción aún mayor de precursores.
La anatomía típica de un paciente con HSC 46,XX incluye grados variables de hipertrofia clitorídea y formación de seno urogenital. Las gónadas son ovarios y no son palpables. Existen varias mutaciones que conducen a distintos perfiles de esteroides suprarrenales característicos de manual y a fenotipos físicos y fisiológicos. La forma más común es la deficiencia de 21-hidroxilasa. Los lactantes con HSC 46,XX pueden presentar pérdida salina potencialmente mortal alrededor de la primera semana de vida, por lo que este diagnóstico debe considerarse en todos los lactantes con genitales ambiguos. Un nivel elevado de 17-OH-progesterona es diagnóstico. La identidad de género femenina y el sexo de crianza femenino son lo más frecuente, aunque no universal.
46,XY DSD
Dentro de la categoría de DSD 46,XY, existe una variedad de trastornos del desarrollo gonadal y de la síntesis y acción de andrógenos, como se detalla en Tabla 3. Junto con el espectro de afecciones de SIA detalladas arriba, la disgenesia gonadal completa o parcial puede dar lugar a variaciones anatómicas similares: un lactante con cromosomas 46,XY y fenotipo femenino típico o ambiguo (p. ej., hipospadias proximal con transposición penoescrotal, y una o ambas gónadas no descendidas). Las personas con SIA completa y disgenesia gonadal completa suelen criarse como mujeres y se identifican como mujeres; aquellas con formas parciales de cualquiera de las dos afecciones presentan mayor variabilidad en el sexo de crianza y la identidad de género.
Otro DSD 46,XY, la deficiencia de 5α-reductasa, provoca una conversión reducida de testosterona a dihidrotestosterona (DHT), lo que da lugar a un fenotipo genital variable. Figura 5 y Figura 6 comparan la vía típica del desarrollo testicular con la vía que se presenta en pacientes con deficiencia de 5α-reductasa.
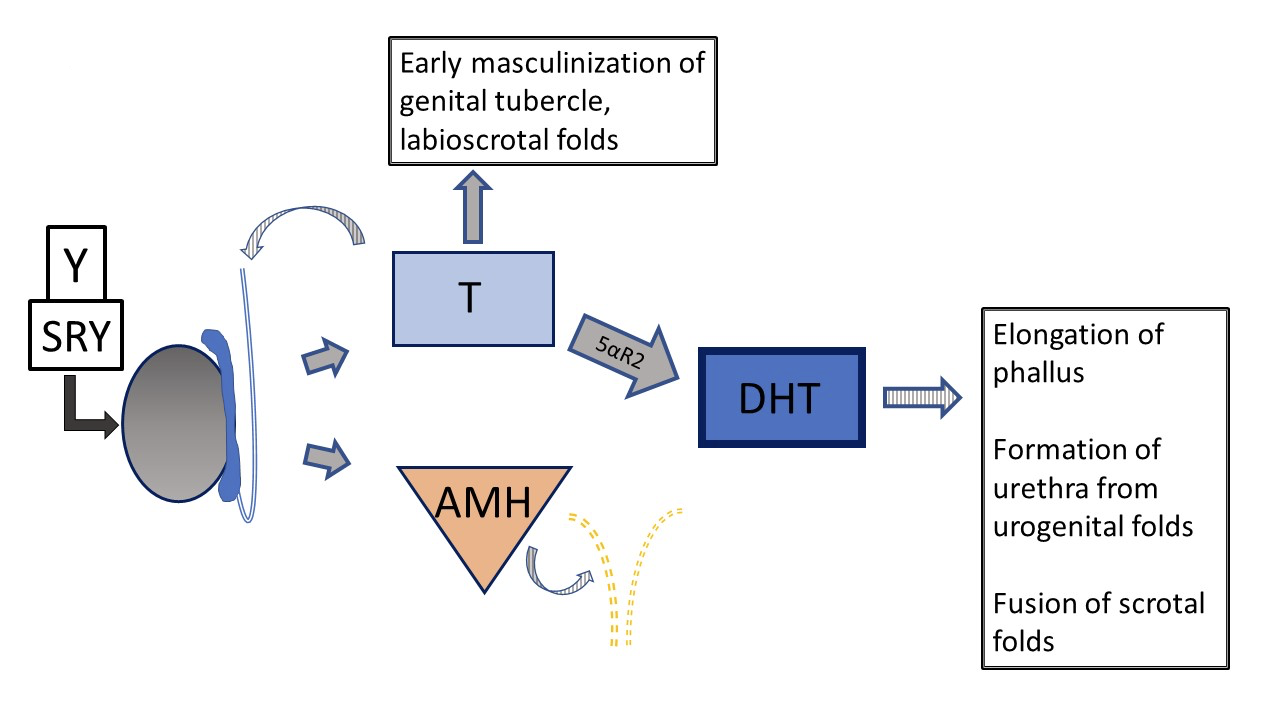
Figura 5 Desarrollo genital prenatal típico 46XY El desarrollo puede desviarse de lo “típico” en varios pasos diferentes. Las flechas a rayas indican una función de tipo paracrino. Las líneas discontinuas indican la regresión de los conductos de Müller en etapas tempranas del desarrollo.
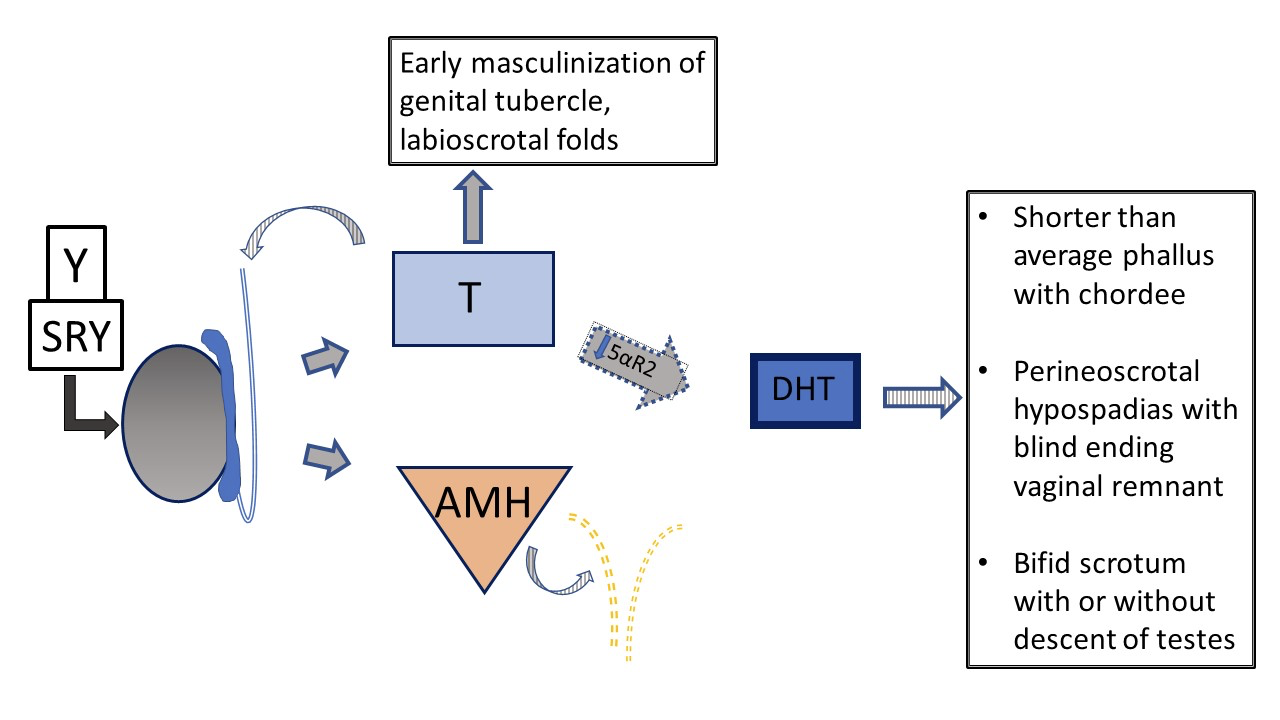
Figura 6 Desarrollo genital prenatal 46XY en pacientes con vs deficiencia de 5-α-reductasa. Los testículos están determinados y presentan una producción normal de testosterona y AMH. No hay útero. Debido a la función deteriorada de la 5-α-reductasa, la relación T /DHT está notablemente aumentada. En el contexto de niveles bajos de DHT, existe variabilidad en el fenotipo desde femenino típico hasta masculino típico, pasando por anatomía genital ambigua. Con una deficiencia grave, el desarrollo del falo y del seno urogenital se detiene en una etapa temprana de la masculinización, con un único orificio urogenital en la región perineal y sin fusión de los pliegues labioscrotales.
Figura 7 muestra la cascada suprarrenal resultante en el ejemplo poco frecuente de un paciente con 46,XY CAH que se presenta clínicamente con genitales ambiguos: deficiencia de 3-β-hidroxiesteroide deshidrogenasa [HSD]. El bloqueo enzimático ocurre antes de la conversión de dehidroepiandrosterona (DHEA) a androstenediona en la cascada de esteroides suprarrenales, por lo que los lactantes presentan niveles reducidos de mineralocorticoides y glucocorticoides junto con una diferencia en el desarrollo genital. Al igual que los bebés con 46,XX CAH y deficiencia de 21-hidroxilasa o 11β-hidroxilasa, los lactantes con deficiencia de 3-β-HSD pueden presentar pérdida de sal potencialmente mortal. El diagnóstico se establece mediante elevación de 17-OH pregnenolona y DHEA.
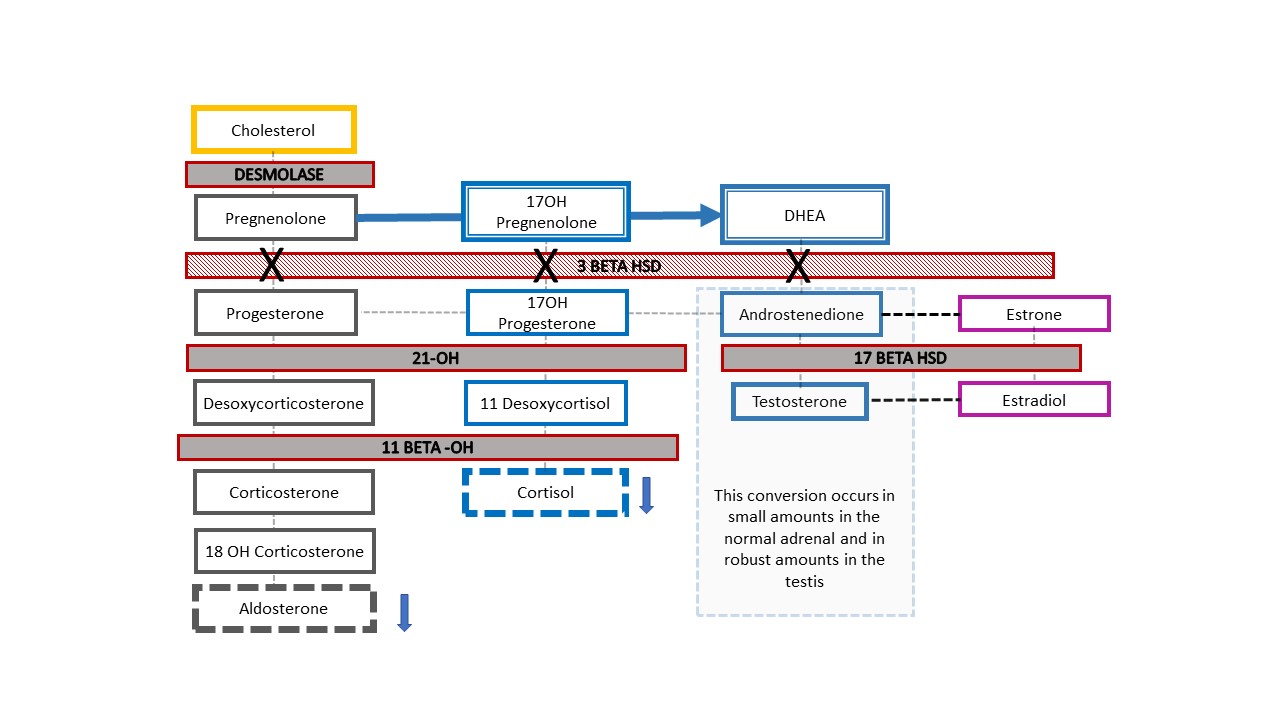
Figura 7 Vía esteroidea en la deficiencia de 3‑β‑hidroxiesteroide deshidrogenasa. Este defecto relativamente proximal en la cascada del colesterol suprarrenal da lugar a una deficiencia grave de corticosteroides y mineralocorticoides. Es única, ya que los bebés 46,XY con HSC presentan una apariencia genital infra-masculinizada, mientras que los bebés 46,XX tienen una apariencia genital femenina típica (o casi típica). Cuando un recién nacido con cromosoma Y detectado prenatalmente nace con una apariencia ambigua de los genitales, este es el diagnóstico de HSC que hay que descartar. La elevación de 17‑OH‑pregnenolona y DHEA ayuda a establecer el diagnóstico.
Trastornos del desarrollo sexual por cromosomas sexuales
De los diagnósticos de DSD de los cromosomas sexuales enumerados en Tabla 3, las afecciones con un cariotipo en mosaico 45,X/46,XY (o similar) merecen mención especial. Los pacientes con este cariotipo pueden presentar genitales externos típicamente femeninos (síndrome de Turner con material del cromosoma Y [TS+Y]), genitales ambiguos (disgenesia gonadal mixta [MGD]) o genitales externos típicamente masculinos. Los pacientes con TS+Y presentan un mayor riesgo de tumores gonadales, a diferencia de los pacientes con síndrome de Turner 45,X. El fenotipo y el sexo de crianza/la identidad de género son variables en la MGD, aunque el resultado es con frecuencia asimetría gonadal y labioescrotal, como se detalla en Figura 8. Cualquier paciente con cariotipo 45,X/46,XY puede presentar manifestaciones del TS (p. ej., coartación aórtica, anomalías renales), por lo que debe someterse a las mismas pruebas de cribado sistémico que los pacientes con TS clásico.7,8
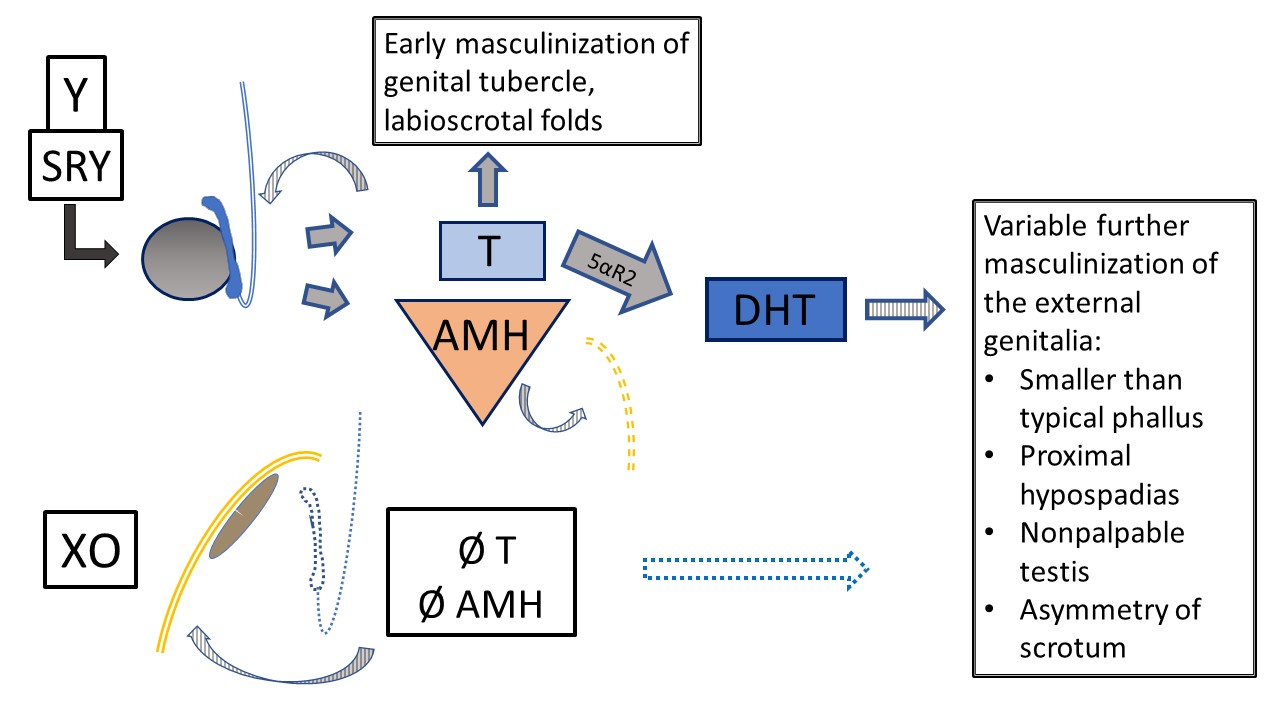
Figura 8 45,X/46,XY Disgenesia gonadal mixta. Se presentan múltiples fenotipos con este cariotipo en mosaico. Este diagrama representa el desarrollo interno y externo en el contexto de un testículo (determinado por el cromosoma Y) y una gónada en banda (desarrollo por defecto, estroma de tipo ovárico). El fenotipo clásico, cuando se observa cierto grado de ambigüedad, es una gónada no palpable, un testículo descendido y un hipospadias proximal. Dado que la T y la AMH tienen función paracrina, la gónada en banda se asocia con un hemiútero y una trompa de Falopio, sin estructuras de Wolff. Debido al material del cromosoma Y, existe riesgo de gonadoblastoma y disgerminoma. El testículo presenta un desarrollo disgenético variable que afecta la función temprana y futura y el riesgo tumoral. Se produce masculinización primaria, pero el desarrollo ulterior de la uretra y el escroto se ve afectado de forma variable por la función testicular.
DSD ovotesticular
El DSD ovotesticular ocurre cuando hay tanto tejido ovárico como testicular presentes en el mismo individuo (nombre anterior – hermafroditismo verdadero). Por lo general, el tejido ovárico está mejor desarrollado y el tejido testicular es disgenético. El fenotipo, el sexo de crianza/identidad de género y el potencial de fertilidad son variables. Es importante señalar que los pacientes con DSD ovotesticular pueden tener cualquier cariotipo (aunque 46, XX es el más frecuente), por lo que debe considerarse este diagnóstico en cualquier paciente que se presente con genitales ambiguos.
Evaluación multidisciplinaria
Más allá de proponer una nomenclatura menos peyorativa, otros pilares del documento de consenso de 2006, en cuanto al paradigma, son el asesoramiento por un equipo multidisciplinario y el apoyo psicosocial longitudinal para pacientes y padres.1
El equipo multidisciplinario
El equipo que colabora con la familia en la interconsulta hospitalaria del recién nacido es, preferentemente, el mismo que trabajará con los pacientes y las familias de manera longitudinal. El equipo es responsable de educar a la familia sobre la afección y todas las posibles opciones de manejo; proporciona apoyo e involucra a la familia en la toma de decisiones compartida cuando se requieren decisiones (p. ej., sexo de crianza, divulgación, cirugía). Idealmente, el equipo incluye los siguientes miembros:
- Endocrinología
- Salud mental (psicólogo, trabajador social)
- Urología / Cirugía pediátrica / Ginecología
- Consejero genético / genetista
- Enfermería
- Defensor del paciente / educador del paciente
- Neonatólogo (interconsulta hospitalaria)
Cada especialista aporta diferentes perspectivas, experiencia clínica relevante y habilidades interpersonales al equipo multidisciplinario. Los cirujanos son una parte importante del equipo, a pesar de que las necesidades quirúrgicas precoces o urgentes sean poco frecuentes. Todos participan en las discusiones del equipo y contribuyen a la experiencia del paciente y la familia. Idealmente, el respeto mutuo y la disposición a considerar todas las perspectivas mejoran el funcionamiento del equipo y permiten que el equipo brinde una atención de un nivel aún más alto con el tiempo, en particular a medida que evolucionan los estándares de atención.
Cada persona que vive con una condición DSD puede no tener acceso a un equipo multidisciplinario funcional. Algunas personas pueden no estar dispuestas a trabajar con el equipo o pueden considerar engorroso el seguimiento longitudinal. Cada oportunidad temprana de educación acerca del diagnóstico y de las posibles preocupaciones o necesidades médicas a largo plazo es, por lo tanto, muy importante. En situaciones en las que el seguimiento a largo plazo sea factible y aceptable, esto puede lograrse en el contexto de una atención más periódica de una sola especialidad, según corresponda al paciente. Por ejemplo, algunos de los pacientes con CAH en la institución de los autores acuden con su endocrinólogo trimestralmente y participan en una visita anual con el equipo multidisciplinario completo.
Trabajar activamente en conjunto, aprender unos de otros y de los pacientes y sus familias, facilita el funcionamiento del equipo e impulsa el avance de la atención. El equipo se apoya en experiencias previas y asimila nuevos conocimientos, en particular en los ámbitos de la experiencia del paciente y de las pruebas genéticas de nueva generación. El equipo también identifica lagunas en el conocimiento y dentro del paradigma de atención que pueden abordarse mediante colaboraciones e investigación deliberada. Las conferencias multidisciplinarias periódicas basadas en casos o en temas ayudan a desarrollar dominio sobre las diversas afecciones entre los miembros del equipo y las personas en formación, y brindan la oportunidad de comprender diferencias de enfoque.
Enfoque inicial del paciente con sospecha de DSD
Los pacientes con sospecha de DSD clásicamente se presentan en la infancia con genitales ambiguos, o en la adolescencia con amenorrea primaria. Las afecciones de DSD también pueden llegar a la atención médica debido a hallazgos incidentales en estudios de imagen o hallazgos intraoperatorios, a una hernia inguinal en una niña, o si una niña experimenta masculinización en el momento de la pubertad. Cada vez con mayor frecuencia, las afecciones de DSD se sospechan de forma prenatal debido a discordancia entre el resultado de ADN libre fetal y la ecografía, o por preocupación ante una diferencia en el desarrollo genital observada en la ecografía.
Cuando nace un recién nacido con sospecha de DSD, se recomienda la consulta con un equipo multidisciplinario (descrito arriba) siempre que sea factible. Recuerde felicitar a la familia por su nuevo bebé (un paso que a menudo se pasa por alto, especialmente cuando hay sorpresa o falta de familiaridad con DSD). Utilice términos neutros en cuanto al género, (“su bebé”, no “ello”) al referirse al bebé si existe alguna duda sobre la asignación de sexo. Figura 9 muestra recomendaciones para describir el examen genital del recién nacido, incluidos los términos neutros en cuanto al género que se deben usar. Junto con una historia prenatal detallada, antecedentes familiares y examen físico, las recomendaciones para la evaluación inicial de un paciente con sospecha de DSD incluyen:
- Cariotipo
- Pruebas endocrinas (17-OH-progesterona, testosterona, hormona luteinizante, hormona estimulante del folículo, electrolitos)
- Ecografía pélvica para evaluar la posición y el tipo de las gónadas, y la presencia de estructuras müllerianas
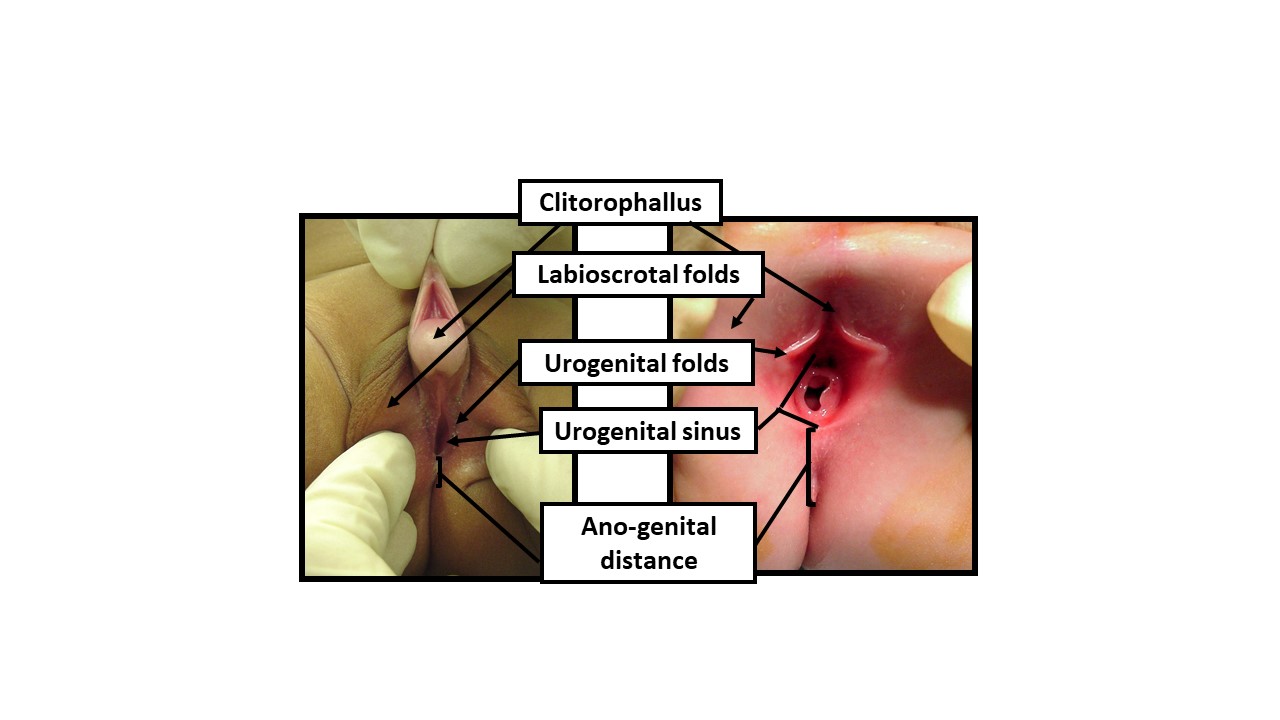
Figura 9 Descripción del examen del recién nacido con términos anatómicos neutros en cuanto al género. Al examinar a un recién nacido con una apariencia ambigua de los genitales, debería utilizarse terminología neutra en cuanto al género hasta que datos suficientes permitan la toma de decisiones compartida con los progenitores para la determinación del sexo de crianza. El examen debería documentar las siguientes manifestaciones de exposición a andrógenos: 1) Longitud de la estructura clitorofálica, 2) Anchura del glande, 3) Número y posición de los orificios perineales, 4) Grado de fusión de los pliegues labioscrotales, 5) Hiperpigmentación y rugosidad de los pliegues labioscrotales, 6) Distancia anogenital, 7) Localización de cualquier gónada palpable
Un enfoque algorítmico basado en el número de gónadas palpables y en los resultados endocrinos, genéticos y de las pruebas de imagen, como se indicó arriba, puede ayudar de forma eficiente a acotar el diagnóstico diferencial. Lo más crítico desde el punto de vista médico en el recién nacido es descartar la CAH perdedora de sal que amenaza la vida. Todo el equipo multidisciplinario descrito anteriormente desempeña un papel durante este período diagnóstico crítico para ayudar a la familia a integrar nueva información y a tomar decisiones sobre el sexo de crianza, la divulgación y la cirugía.
Opciones de manejo quirúrgico
Las opciones quirúrgicas para los pacientes con DSD son altamente individualizadas, y las decisiones sobre el tratamiento quirúrgico idealmente deberían tomarse en el contexto de un equipo multidisciplinario. Según las necesidades y los objetivos del paciente, los procedimientos pueden ser diagnósticos, reconstructivos o extirpativos.
Procedimientos diagnósticos
Suelen realizarse procedimientos diagnósticos cuando la anatomía o el diagnóstico siguen sin estar claros tras la evaluación endocrina y genética y las imágenes radiológicas detalladas.
La endoscopia proporciona una visualización detallada y mediciones del tracto urogenital. Por ejemplo, es posible caracterizar la longitud y la configuración de un seno urogenital. Las estructuras vaginales pueden evaluarse en cuanto a profundidad, número y presencia o ausencia de cuello uterino.
La laparoscopia diagnóstica facilita la visualización de las gónadas y las estructuras müllerianas. Esta visualización puede ayudar en el diagnóstico y aportar información para la planificación quirúrgica reconstructiva futura. La biopsia gonadal puede realizarse por vía laparoscópica o abierta para determinar el tipo de tejido (por ejemplo, si se sospecha un DSD ovotesticular), o enviarse para cariotipo si se sospecha material del cromosoma Y pero no se ha identificado en el cariotipo de sangre periférica. Figura 10 muestra imágenes fijas laparoscópicas de un paciente con MGD: el diagnóstico de MGD se sospechó clínicamente y se confirmó con el aspecto gonadal macroscópico e histológico proporcionado por la laparoscopia diagnóstica y la biopsia.
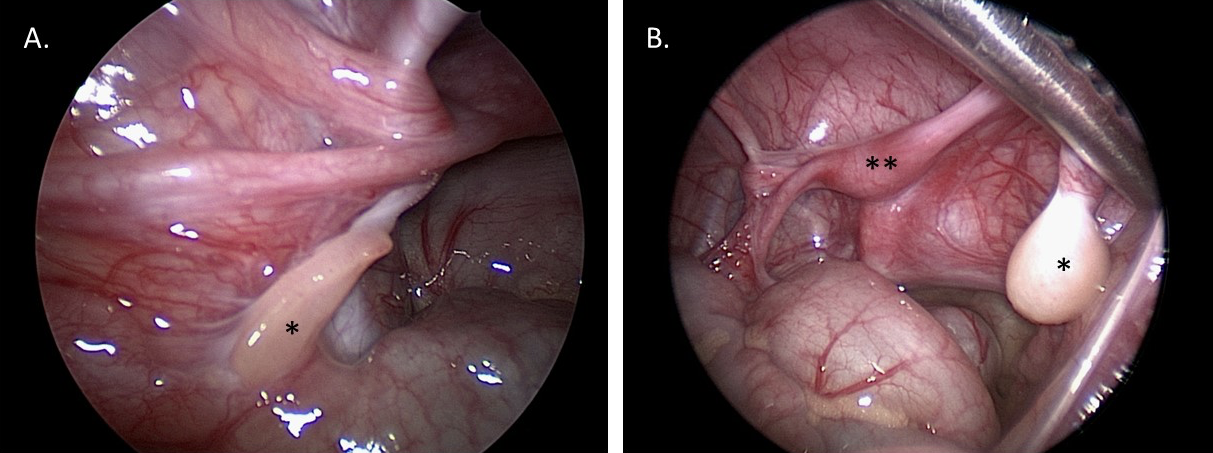
Figura 10 Imágenes laparoscópicas diagnósticas de la pelvis en un paciente de 1 año con disgenesia gonadal mixta. A. *Gónada en banda izquierda. B. *Testículo disgenético derecho. **Útero.
Genitoplastia masculinizante
Para pacientes con DSD, la genitoplastia masculinizante con mayor frecuencia implica una reparación de hipospadias proximal, a menudo con escrotoplastia para corregir la transposición penoescrotal y/o una configuración bífida. Los detalles técnicos de la reparación del hipospadias son similares a los de los pacientes sin un DSD definido. La reparación del hipospadias en pacientes con DSD típicamente requiere un procedimiento en dos etapas para lograr la corrección de la curvatura peneana y un meato uretral terminal. Existe controversia en la literatura acerca de si los pacientes con un diagnóstico conocido de DSD tienen peores resultados quirúrgicos del hipospadias en comparación con los pacientes con hipospadias proximal que no tienen un diagnóstico específico de DSD.9,10,11
Genitoplastia feminizante
Los componentes incluyen clitoroplastia (con labioplastia) y vaginoplastia. Existe una variedad de abordajes, que pueden adaptarse según la anatomía y los objetivos de la paciente. Un ejemplo de marcaje quirúrgico preoperatorio para una paciente con HSC que se sometió a una movilización urogenital parcial (PUM) y labioplastia sin clitoroplastia se ilustra en Figura 11.
Clitoroplastia y Labioplastia
Los procedimientos modernos de clitoroplastia reportados con mayor frecuencia implican una reducción del clítoris con preservación nerviosa.12 El clítoris se desenfunda y se extirpa tejido corporal (eréctil), evitando los haces neurovasculares dorsales. La técnica de clitoroplastia de reducción con preservación de la túnica albugínea se introdujo para preservar aún más el aporte sanguíneo y la sensibilidad del glande, e implica incisiones corporales ventrales y escisión del tejido eréctil, preservando la túnica circundante. El glande del clítoris se preserva con frecuencia, se reubica proximalmente y se fija por debajo del pubis. La reducción del glande puede realizarse mediante la escisión de una cuña ventral de tejido y la reaproximación de los bordes ventrales entre sí. Sin embargo, este abordaje conlleva el riesgo de una disminución de la sensibilidad del glande. Con la clitoroplastia junto con la labioplastia, a menudo se puede lograr un clítoris oculto, por lo que la reducción del glande es cada vez menos frecuente.
Han surgido varias técnicas que preservan los cuerpos cavernosos como alternativas al procedimiento de reducción clitoriana descrito anteriormente. Como ejemplo, Pippi Salle describió un procedimiento de desmontaje corporal.13 En este procedimiento, los cuerpos cavernosos se separan completamente del glande y del paquete neurovascular, y entre sí. Luego, los cuerpos cavernosos se tunelizan individualmente dentro de sus respectivos labios mayores ipsilaterales, y el glande se reposiciona proximalmente, de manera similar a los procedimientos de reducción clitoriana. Este ‘procedimiento de Pippi Salle’ tiene la ventaja teórica de ser reversible, ya que no se extirpa tejido, aunque no se han reportado intentos de reversión ni otros resultados a largo plazo.
La labioplastia es típicamente el paso final de un procedimiento de clitoroplastia (con o sin vaginoplastia). Los objetivos son reconfigurar el prepucio clitorídeo y el tejido del cuerpo del clítoris sobre el clítoris para cubrir el glande clitorídeo, y avanzar los bordes cutáneos hacia posterior, laterales al vestíbulo/introito vaginal.
Vaginoplastia
El enfoque de la vaginoplastia implica una evaluación para determinar si la paciente presenta un seno urogenital con confluencia baja (canal común corto con separación de la vagina y la uretra cercana al periné) o confluencia alta (canal común más largo y mayor separación.14
Una vaginoplastia con colgajo cutáneo perineal (técnica de Fortunoff) puede ser suficiente para pacientes con un seno urogenital de confluencia baja. Para esta técnica, se crea un colgajo posterior con forma de U (como en Figura 11) A continuación, se incide la pared posterior de la vagina proximal a la confluencia displásica, y el colgajo se posiciona y fija dentro de esta incisión vaginal para aumentar el calibre vaginal.
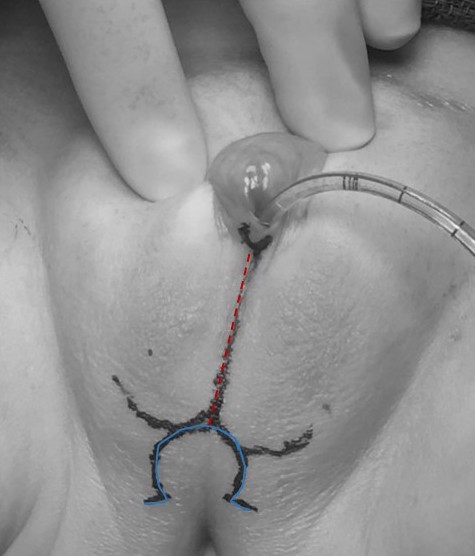
Figura 11 Marcaje quirúrgico preoperatorio en una paciente con hiperplasia suprarrenal congénita que será sometida a una movilización urogenital parcial y labioplastia sin clitoroplastia. Se observa un catéter dentro del orificio del seno urogenital. La marca de la línea media (rojo discontinuo) se extiende a través de los pliegues labioescrotales fusionados, y la marca en forma de omega (línea azul continua) formará el colgajo cutáneo posterior para la vaginoplastia.
En la movilización urogenital total (TUM) o parcial (PUM), la uretra y la vagina se movilizan hacia el periné como una unidad.15 La distinción entre ambas radica en si la disección anterior por detrás del pubis secciona el ligamento pubouretral, como se muestra en la Figura 12. Existe la posible preocupación por incontinencia urinaria con una movilización más extensa, pero esto no se ha demostrado de manera concluyente y probablemente sea más preocupante si la distancia desde la confluencia hasta el cuello vesical está acortada. Si se aplica a casos de confluencia baja, la PUM generalmente lleva la vagina a una posición en la que puede exteriorizarse o ser puenteada con un colgajo perineal de base posterior (ver arriba). La confluencia alta implica una anatomía más compleja, que incluye una mayor distancia para alcanzar el periné, una relación más estrecha entre la vagina y el cuello vesical y, con frecuencia, una vagina más corta. El descenso directo puede no ser ventajoso a largo plazo. En los casos de confluencia alta, incluso con la movilización urogenital, será necesario separar la vagina del seno urogenital y descenderla hasta el periné. En este escenario, el seno urogenital se convierte en la uretra. La vagina alcanza el periné con la ayuda de colgajos cutáneos perineales o utilizando tejido excedente del seno urogenital movilizado como colgajo.
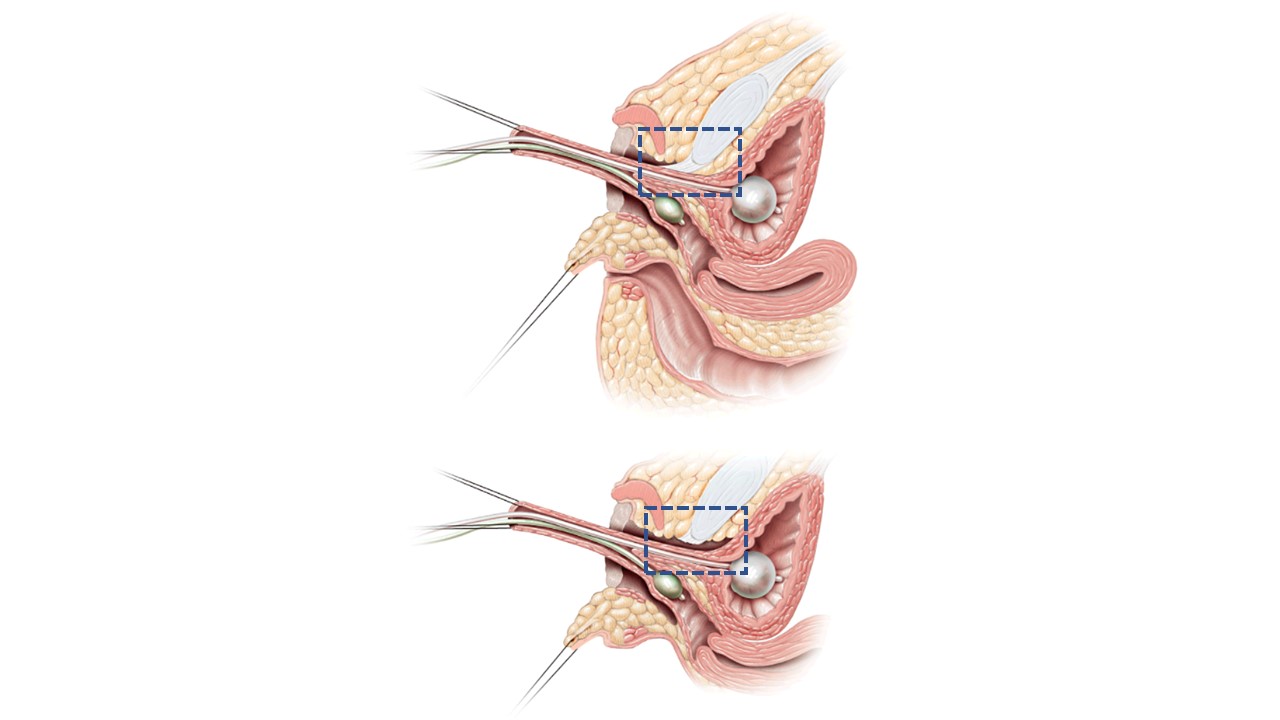
Figura 12 Imágenes de corte sagital de movilización urogenital parcial vs total. A. Movilización urogenital parcial: la uretra y la vagina se avanzan hacia el periné como una unidad. El ligamento pubouretral (recuadro de línea discontinua azul) se deja intacto. B. Movilización urogenital total: la uretra y la vagina se avanzan hacia el periné como una unidad. El ligamento pubouretral (recuadro de línea discontinua azul) se divide. Adaptado de: Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int 2008; 102 (9): 1182–1197. DOI: 10.1111/j.1464-410x.2008.08091.x. (permiso concedido 2/1/2022)
En pacientes que nacen con estructuras müllerianas ausentes o poco desarrolladas, la vaginoplastia consiste en la creación mediante disección roma de una neovagina en el espacio retrovesical entre la vejiga y el recto, y luego en el revestimiento del nuevo espacio potencial con tejido autólogo (p. ej., injerto cutáneo, injerto de mucosa bucal, colgajo peritoneal) u otros injertos biológicos (p. ej., submucosa del intestino delgado [SIS]). Estas técnicas también pueden utilizarse como complemento de los procedimientos descritos arriba, o en el contexto de reoperación.
Dilataciones vaginales
Algunas personas con DSD (p. ej., AIS completa) pueden lograr una vagina que permita una actividad sexual cómoda y placentera con dilataciones vaginales regulares, sin necesidad de una vaginoplastia quirúrgica. Todas las personas que se someten a una vaginoplastia quirúrgica también requieren dilataciones vaginales mientras se recuperan de la cirugía, y hasta que tengan relaciones sexuales vaginales de forma regular.
Gonadectomía y manejo gonadal
El paradigma del manejo gonadal en pacientes con DSD ha evolucionado con el tiempo. Tradicionalmente, a los pacientes con DSD en los que existe un mayor riesgo de formación tumoral se les aconsejaba someterse a gonadectomía. Se asumía la infertilidad. Con el tiempo, se ha hecho evidente que existe un amplio rango de riesgo tumoral y que los tumores que se forman son con frecuencia gonadoblastomas (no malignos).16,17 Los disgerminomas (tumores malignos de células germinales) son posibles, pero sumamente raros antes de la pubertad. También puede existir un potencial de fertilidad no tradicional en algunas personas previamente consideradas infértiles.18 Por lo tanto, las recomendaciones de manejo gonadal se están volviendo más individualizadas, considerando el diagnóstico del paciente (y, por ende, el riesgo tumoral previsto), la edad y el potencial de función gonadal endógena (tanto hormonal como reproductiva). Para los pacientes que optan por la gonadectomía, la criopreservación de tejido gonadal es una opción emergente que puede posibilitar la fertilidad biológica, suponiendo que se produzcan los avances futuros esperados en las tecnologías de reproducción asistida.19 Aún no existen protocolos de vigilancia basados en evidencia para los pacientes que optan por conservar sus gónadas; la institución de los autores suele recomendar ecografía pélvica cada 6–12 meses. Tabla 4 muestra estrategias de recomendación gonadal para varios diagnósticos de DSD.
Tabla 4 Opciones de manejo gonadal según el diagnóstico. * considerar la criopreservación experimental del tejido gonadal.
| Diagnóstico | Riesgo tumoral estimado | ¿Se esperan células germinales? | ¿Se espera pubertad? | Opciones de manejo de las gónadas |
|---|---|---|---|---|
| 46, XY Disgenesia gonadal completa (síndrome de Swyer) | Alto – Hasta un 35% |
No (quizá células germinales precursoras) |
No | -Gonadectomía* |
| Síndrome de insensibilidad parcial a los andrógenos | Alto – Hasta un 50% según la localización de la gónada |
Sí Posibles espermatozoides |
Sí, variable | -Orquidopexia, considerar biopsia -Observación con ecografías -Gonadectomía* |
| Síndrome de Turner + cromosoma Y | Medio – ~10-30% |
Quizá – Posibles ovocitos | Posible | -Observación con ecografías -Gonadectomía* (estándar, bajo nivel de evidencia) |
| Síndrome de insensibilidad completa a los andrógenos | Bajo – ~2-10% |
Sí – Precursores de espermatozoides |
Sí – debido a la aromatización de la testosterona a estrógenos | -Gonadectomía pospuberal* -Observación con ecografías, RM |
Manejo de las estructuras müllerianas
En los pacientes que están siendo criados como varón y presentan estructuras müllerianas persistentes (p. ej., un utrículo grande), por lo general se dejan en su lugar salvo que se vuelvan sintomáticas. La exéresis conlleva el riesgo potencial de lesión de las estructuras deferenciales (y posiblemente de los nervios pélvicos), y los pacientes con frecuencia están asintomáticos. La reparación del hipospadias es segura, y la exéresis de estructuras utriculares incluso grandes no es obligatoria antes de la reparación del hipospadias. Si aparecen síntomas como infecciones o hematuria cíclica molesta, puede plantearse la exéresis quirúrgica mediante un abordaje abierto, laparoscópico o robótico. También puede considerarse el manejo hormonal con supresión menstrual para los pacientes que experimentan dolor cíclico debido a un remanente mülleriano obstruido, o hematuria cíclica molesta.
Conclusiones
Los DSD abarcan una amplia gama de afecciones que resultan de un desarrollo cromosómico, gonadal o anatómico anormal. Existe una amplia gama de fenotipos, incluso dentro de diagnósticos y categorías específicas de DSD. Siempre que sea posible, se recomienda un enfoque multidisciplinario para la evaluación y el seguimiento a largo plazo.
Puntos clave
- La terminología de DSD aceptada en el ámbito médico no es la preferida por todas las personas afectadas ni por sus familias. En los encuentros clínicos individuales, pregunte a los pacientes y a las familias qué términos prefieren usar al referirse al diagnóstico y a la anatomía.
- Las afecciones DSD ocurren debido a cromosomas, gónadas o acción hormonal atípicos. Considerar el grado de anomalía y determinar si el defecto identificado es completo o parcial puede ayudar a predecir el fenotipo del paciente.
- El número de gónadas palpables sienta las bases para un diagnóstico diferencial de DSD. El diagnóstico diferencial se refina aún más mediante cariotipo, pruebas de laboratorio endocrinas y ecografía pélvica.
- Recuerde felicitar a los nuevos padres que tienen un lactante con sospecha de DSD—este paso crítico a menudo se olvida.
- CAH es la forma más común de DSD 46, XX. Las características clínicas incluyen genitales externos masculinizados con gónadas no palpables. La pérdida salina potencialmente mortal puede ocurrir en la primera semana de vida.
- El DSD ovotesticular puede presentarse en un paciente con cualquier cariotipo.
- Un paciente con un cariotipo 45, X/46, XY puede tener genitales externos típicamente femeninos, ambiguos o típicamente masculinos. Cualquier paciente con este cariotipo puede presentar manifestaciones sistémicas del síndrome de Turner.
Lecturas recomendadas
- Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
- Kaefer M, Diamond D, Hendren WH. The Incidence Of Intersexuality In Children With Cryptorchidism And Hypospadias: Stratification Based On Gonadal Palpability And Meatal Position. J Urol 1999; 162 (3 Part 2): 1006–1007. DOI: 10.1016/s0022-5347(01)68049-2.
- Kim S, Rosoklija I, Johnson EK. Surgical, Patient, and Parental Considerations in the Management of Children with Differences of Sex Development. Curr Pediatr Rep 2018; 6 (3): 209–219. DOI: 10.1007/s40124-018-0177-4.
- Mouriquand PD, Gorduza DB, Gay CL. Re: “Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how?” J Pediatr Urol 2016; 12 (6): 442–443. DOI: 10.1016/j.jpurol.2016.07.013.
- Nixon R, Cerqueira V, Kyriakou A, Lucas-Herald A, McNeilly J, McMillan M, et al.. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod 2017; 32 (10): 2130–2137. DOI: 10.1093/humrep/dex280.
- Van Batavia JP, Kolon TF. Fertility in disorders of sex development: A review. J Pediatr Urol 2016; 12 (6): 418–425. DOI: 10.1016/j.jpurol.2016.09.015.
Referencias
- Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
- Johnson EK, Rosoklija I, Finlayson C. Faculty Opinions recommendation of Attitudes towards "disorders of sex development" nomenclature among affected individuals. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2017; 13: 608. DOI: 10.3410/f.727655647.793533819.
- D’Oro A, Rosoklija I, Jacobson DL, Finlayson C, Chen D, Tu DD, et al.. Patient and Caregiver Attitudes toward Disorders of Sex Development Nomenclature. J Urol 2020; 204 (4): 835–842. DOI: 10.1097/ju.0000000000001076.
- Davies JH, Knight EJ, Savage A, Brown J, Malone PS. Evaluation of terminology used to describe disorders of sex development. J Pediatr Urol 2011; 7 (4): 412–415. DOI: 10.1016/j.jpurol.2010.07.004.
- Lin-Su K, Lekarev O, Poppas DP, Vogiatzi MG. Congenital adrenal hyperplasia patient perception of ‘disorders of sex development’ nomenclature. Int J Pediatr Endocrinol 2015; 2015 (1): 9. DOI: 10.1186/s13633-015-0004-4.
- Snodgrass W, Macedo A, Hoebeke P, Mouriquand PDE. Hypospadias dilemmas: A round table. J Pediatr Urol 2011; 7 (2): 145–157. DOI: 10.1016/j.jpurol.2010.11.009.
- Wu Q, Wang C, Shi H, Kong X, Ren S, Jiang M. The Clinical Manifestation and Genetic Evaluation in Patients with 45,X/46,XY Mosaicism. Sex Dev 2017; 11 (2): 64–69. DOI: 10.1159/000455260.
- Das DV, Jabbar PK. Clinical and Reproductive Characteristics of Patients with Mixed Gonadal Dysgenesis (45,X/46, XY). J Obstet Gynaecol India 2021; 71 (4): 399–405. DOI: 10.1007/s13224-021-01448-3.
- Saltzman AF, Carrasco A, Colvin A, Campbell JB, Vemulakonda VM, Wilcox D. Patients with disorders of sex development and proximal hypospadias are at high risk for reoperation. World J Urol 2018; 36 (12): 2051–2058. DOI: 10.1007/s00345-018-2350-3.
- Ochi T, Ishiyama A, Yazaki Y, Murakami H, Takeda M, Seo S, et al.. Surgical management of hypospadias in cases with concomitant disorders of sex development. Pediatr Surg Int 2019; 35 (5): 611–617. DOI: 10.1007/s00383-019-04457-6.
- Palmer BW, Reiner W, Kropp BP. Proximal Hypospadias Repair Outcomes in Patients with a Specific Disorder of Sexual Development Diagnosis. Adv Urol 2012; 2012: 1–4. DOI: 10.1155/2012/708301.
- Kaefer M, Rink RC. Treatment of the Enlarged Clitoris. Front Pediatr 2017; 5. DOI: 10.3389/fped.2017.00125.
- Pippi Salle JL, Braga LP, Macedo N, Rosito N, Bagli D. Corporeal Sparing Dismembered Clitoroplasty: An Alternative Technique for Feminizing Genitoplasty. J Urol 2007; 178 (4s): 1796–1801. DOI: 10.1016/j.juro.2007.03.167.
- Guarino N, Scommegna S, Majore S, Rapone AM, Ungaro L, Morrone A, et al.. Vaginoplasty for Disorders of Sex Development. Front Endocrinol (Lausanne) 2013; 4: 29, DOI: 10.3389/fendo.2013.00029.
- Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int 2008; 102 (9): 1182–1197. DOI: 10.1111/j.1464-410x.2008.08091.x.
- Looijenga LHJ, Hersmus R, Oosterhuis JW, Cools M, Drop SLS, Wolffenbuttel KP. Tumor risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab 2007; 21 (3): 480–495. DOI: 10.1016/j.beem.2007.05.001.
- Looijenga LHJ, Hersmus R, Leeuw BHCGM de, Stoop H, Cools M, Oosterhuis JW, et al.. Gonadal tumours and DSD. Best Pract Res Clin Endocrinol Metab 2010; 24 (2): 291–310. DOI: 10.1016/j.beem.2009.10.002.
- Finlayson C, Fritsch MK, Johnson EK, Rosoklija I, Gosiengfiao Y, Yerkes E, et al.. Presence of Germ Cells in Disorders of Sex Development: Implications for Fertility Potential and Preservation. J Urol 2017; 197 (3 Part 2): 937–943. DOI: 10.1016/j.juro.2016.08.108.
- Harris CJ, Corkum KS, Finlayson C, Rowell EE, Laronda MM, Reimann MB, et al.. Establishing an Institutional Gonadal Tissue Cryopreservation Protocol for Patients with Differences of Sex Development. J Urol 2020; 204 (5): 1054–1061. DOI: 10.1097/ju.0000000000001128.
- Kaefer M, Diamond D, Hendren WH. The Incidence Of Intersexuality In Children With Cryptorchidism And Hypospadias: Stratification Based On Gonadal Palpability And Meatal Position. J Urol 1999; 162 (3 Part 2): 1006–1007. DOI: 10.1016/s0022-5347(01)68049-2.
- Kim S, Rosoklija I, Johnson EK. Surgical, Patient, and Parental Considerations in the Management of Children with Differences of Sex Development. Curr Pediatr Rep 2018; 6 (3): 209–219. DOI: 10.1007/s40124-018-0177-4.
- Mouriquand PD, Gorduza DB, Gay CL. Re: “Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how?” J Pediatr Urol 2016; 12 (6): 442–443. DOI: 10.1016/j.jpurol.2016.07.013.
- Nixon R, Cerqueira V, Kyriakou A, Lucas-Herald A, McNeilly J, McMillan M, et al.. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod 2017; 32 (10): 2130–2137. DOI: 10.1093/humrep/dex280.
- Van Batavia JP, Kolon TF. Fertility in disorders of sex development: A review. J Pediatr Urol 2016; 12 (6): 418–425. DOI: 10.1016/j.jpurol.2016.09.015.
Última actualización: 2025-09-21 13:35