
13: Enfermedades quísticas del riñón
Este capítulo durará aproximadamente 24 minutos para leer.
Introducción
Las enfermedades renales quísticas constituyen una categoría amplia de afecciones renales esporádicas y genéticas, congénitas o adquiridas, que pueden implicar la presencia de un único quiste hasta múltiples quistes en uno o ambos riñones (véanse las clasificaciones hereditarias en Tabla 1 más abajo).1 Según el Comité de Clasificación, Nomenclatura y Terminología de 1987, la clasificación de la enfermedad renal quística se determina principalmente por la etiología genética y no genética.2 Las enfermedades renales quísticas pueden considerarse en gran medida afecciones poco frecuentes; sin embargo, pueden asociarse a consecuencias graves, como enfermedad renal crónica (ERC), insuficiencia renal o enfermedad hepática. A pesar de su rareza, la enfermedad renal poliquística autosómica dominante (ADPKD) es una de las afecciones hereditarias más frecuentes en el ser humano. La posibilidad de complicaciones graves derivadas de las enfermedades renales quísticas en la población pediátrica, en especial la ERC, hace que el diagnóstico precoz y las modalidades de tratamiento eficaces sean de particular importancia.
Tabla 1 Clasificaciones de las enfermedades renales quísticas hereditarias.1
| Hereditaria | No hereditaria |
|---|---|
| Enfermedad poliquística renal autosómica recesiva (infantil) | Riñón multiquístico (riñón displásico multiquístico) |
| Enfermedad poliquística renal autosómica dominante (del adulto) | Quiste multilocular benigno (nefroma quístico) |
| Complejo de nefronoftisis juvenil y enfermedad quística medular | Quistes simples |
| Nefronoftisis juvenil (autosómica recesiva) | Riñón en esponja medular |
| Enfermedad quística medular (autosómica dominante) | Enfermedad renal glomeruloquística esporádica |
| Nefrosis congénita (síndrome nefrótico familiar) (autosómica recesiva) | Enfermedad renal quística adquirida |
| Enfermedad glomeruloquística hipoplásica familiar (autosómica dominante) | Divertículo calicial (quiste pielógeno) |
| Síndromes de malformaciones múltiples con quistes renales (p. ej., esclerosis tuberosa, enfermedad de von Hippel-Lindau) |
Embriología
Los quistes suelen ocurrir secundariamente a desequilibrios secretorios, absortivos y electrolíticos a nivel de las células epiteliales y pueden formarse en cualquier parte del sistema tubular renal. Investigaciones recientes sobre la enfermedad renal quística hereditaria sugieren que los genes afectados involucran el cilio primario de las células epiteliales renales, una posible vía común para estas afecciones.3 El término “ciliopatía” se refiere a esta vía común. Los cilios cumplen la función de modular la proliferación y la diferenciación de las células tubulares, y su disfunción puede conducir a una expansión inapropiada de los túbulos y a la formación de quistes.4
Este capítulo analizará la epidemiología, la patogénesis, la evaluación y el tratamiento de diversas enfermedades renales quísticas hereditarias y no hereditarias, incluyendo ADPKD, enfermedad renal poliquística autosómica recesiva (ARPKD), quistes renales aislados, divertículos caliciales, riñón displásico multiquístico (MCDK), riñón en esponja medular, nefronoptisis, quistes multiloculares (nefroma quístico multilocular), enfermedades renales quísticas adquiridas y síndromes asociados con la enfermedad renal quística.
Enfermedad renal poliquística autosómica dominante
ADPKD es relativamente común, con una incidencia de 1 en 400 a 1,000 nacidos vivos que se manifiesta como dilatación quística del nefrón y otras anomalías extrarrenales, como aneurisma intracraneal.5 La gravedad varía según el paciente y tiende a empeorar con el tiempo, por lo que muchos pacientes pediátricos e incluso algunos adultos de mediana edad pueden ser asintomáticos. Aunque poco frecuente, la enfermedad grave en la infancia puede presagiar una morbilidad y mortalidad significativas.6 Debido a los antecedentes familiares o a hallazgos incidentales en estudios de imagen, se están identificando más niños asintomáticos con ADPKD.
Epidemiología
De los afectados con ADPKD, alrededor del 96% presentarán signos clínicos de la enfermedad para los 90 años.7 A pesar de que la mayoría de los casos se descubren en pacientes de mediana edad (30–40 años) y de que la insuficiencia renal antes de los 40 años de edad es rara, se ha descrito tan temprano como en el periodo neonatal. Cuando se describe en recién nacidos, se considera que la ADPKD es más agresiva.8 A pesar del patrón de herencia autosómico dominante con una penetrancia teórica del 100%, el 10% de los casos puede ocurrir de forma esporádica.1
Patogénesis
La ADPKD se hereda de forma autosómica dominante y las principales mutaciones genéticas ocurren en PKD1 y PKD2 en el cromosoma 16 y el cromosoma 4, respectivamente.5 El producto génico es una proteína transmembrana (policistina 1 y policistina 2, respectivamente) localizada dentro de los cilios primarios.9 Las mutaciones de PKD1 en el cromosoma 16 representan la mayoría de los casos y se asocian con mayor gravedad, mientras que la mayoría de los casos restantes se deben a mutaciones de PKD2.10
El funcionamiento anómalo de la policistina 1 o la policistina 2 conduce a la desregulación de las señales de las vías proliferativas, como cAMP, ERK y mTOR. Los cilios pueden desempeñar un papel organizativo en la transducción de estas señales, ya que la desregulación conduce a la formación de quistes.11
Evaluación y diagnóstico
Como estudio de imagen costo-efectivo y mínimamente invasivo, la ultrasonografía renal se utiliza con frecuencia para identificar ADPKD. Los criterios diagnósticos por ultrasonografía en menores de 30 años incluyen la presencia de al menos dos quistes, ya sean unilaterales o bilaterales, requiriéndose un mayor número de quistes para sugerir el diagnóstico a medida que aumenta la edad.12 No obstante, la ADPKD y la ARPKD pueden ser difíciles de diferenciar en la población pediátrica, por lo que la presencia de antecedentes familiares positivos es crucial para el diagnóstico.13 En ausencia de antecedentes familiares, se pueden realizar pruebas genéticas. Otros factores importantes a considerar, cuando los antecedentes familiares son negativos, incluyen aumento bilateral del tamaño renal, tres o más quistes hepáticos, aneurisma de arteria cerebral y quistes solitarios de otros órganos (aracnoides, glándula pineal, páncreas o bazo).1,14 La obtención de imágenes o la biopsia del hígado también puede ayudar a diferenciar ADPKD y ARPKD, ya que la fibrosis hepática está siempre presente en ARPKD, pero es rara en ADPKD.5 Por último, la anatomía patológica renal (biopsia) puede ser útil, ya que la ADPKD puede comprometer la totalidad del túbulo, incluido el glomérulo; sin embargo, la ARPKD no presentaría quistes glomerulares.5
Opciones de tratamiento, resultados y complicaciones
Actualmente no existe cura para la ADPKD. Los objetivos del tratamiento incluyen retrasar el inicio de la enfermedad renal terminal (ERT) y reducir la carga de complicaciones de la ADPKD relacionadas con el deterioro de la función renal, la enfermedad cardíaca y la hemorragia intracraneal. Las complicaciones se modulan de manera más significativa mediante el manejo de la hipertensión.1 Por lo tanto, se recomienda un control riguroso de la presión arterial y los inhibidores de la enzima convertidora de angiotensina (ECA) y los bloqueadores del receptor de angiotensina son una opción común, sin embargo, no existe consenso sobre los antihipertensivos ideales.15,16
Las terapias emergentes en investigación tienen como objetivo limitar el crecimiento de los quistes al reducir el AMPc mediante el uso de inhibidores de mTOR, somatostatina, inhibidores de las quinasas de tirosina, inhibidores de la quinasa Src, inhibidores de MEK e inhibidores de las quinasas dependientes de ciclina.
Enfermedad renal poliquística autosómica recesiva
La ARPKD se caracteriza por un agrandamiento rápido de ambos riñones en lactantes debido a la formación de quistes dentro del sistema de túbulos colectores. Puede asociarse con fibrosis hepática congénita, que puede conducir a hipertensión portal. Una presentación más temprana de la ARPKD suele asociarse con mayor gravedad.1 Sus efectos multisistémicos pueden requerir un manejo multidisciplinario.
Epidemiología
Dado que la ARPKD es menos frecuente que la ADPKD, hay menos datos epidemiológicos. La incidencia reportada es de 1 en 26,500 nacidos vivos, pero puede variar entre 1 en 10,000 y 1 en 50,000 nacidos vivos.1,17 Sin embargo, puede ser más frecuente en poblaciones aisladas o consanguíneas.
Patogénesis
ARPKD se hereda con un patrón autosómico recesivo clásico, con progenitores heterocigotos no afectados que tienen un 25% de riesgo de tener un hijo afectado. Está causada por mutación en el gen de la enfermedad renal y hepática poliquística 1 (PKHD1) del cromosoma 6, que codifica la fibrocistina (también conocida como poliductina), una proteína identificada en el sistema de túbulos renales (conducto colector y rama ascendente gruesa), en las células epiteliales del conducto biliar hepático y en los cilios apicales primarios.5,18,19
Un paciente típico puede presentarse en el período neonatal con antecedentes de oligohidramnios prenatal, riñones aumentados de tamaño y “secuencia de Potter” con hipoplasia pulmonar y muerte perinatal en aproximadamente el 30% de los recién nacidos afectados.20,21 Los riñones aumentados de tamaño aparecen ecogénicos con dilatación fusiforme de los túbulos colectores y la progresión a enfermedad renal terminal (ERT) puede ocurrir a edades variables. El efecto sobre otros sistemas orgánicos puede manifestarse como conductos biliares dilatados, fibrosis hepática congénita, hipertensión portal y disfunción neurocognitiva.22
Evaluación y diagnóstico
La ecografía fetal y del recién nacido puede mostrar riñones bilaterales, muy aumentados de tamaño y difusamente ecogénicos debido a abundantes microquistes.1 La presencia de macroquistes (> 10 mm) puede indicar otras ciliopatías como MCDK o ADPKD y la resonancia magnética puede ayudar a evaluar más a fondo las anomalías en el contexto de oligohidramnios grave.13 Un diagnóstico preciso depende de hallazgos de imagen sugestivos, presencia de antecedentes familiares con patrón de herencia recesivo, biopsia hepática que muestre lesiones periportales (presentes en ARPKD y raras en ADPKD), y ausencia de manifestaciones extrarrenales asociadas con otras enfermedades renales quísticas.1
Opciones de tratamiento, resultados y complicaciones
Al igual que en la ADPKD, no existe cura para la ARPKD, por lo tanto, muchas opciones terapéuticas se orientan al manejo sintomático o la paliación de la hipertensión, la insuficiencia cardíaca congestiva y la insuficiencia renal y hepática. El tratamiento agresivo, como la nefrectomía unilateral o bilateral, puede estar indicado en individuos gravemente afectados que presenten compromiso respiratorio o nutricional debido al efecto de masa de los riñones agrandados. Pueden implementarse terapias descompresivas para el manejo de la hipertensión portal. A menudo se consideran la hemodiálisis y el trasplante renal.1
Quistes renales aislados
Los quistes renales solitarios y múltiples son las lesiones renales más frecuentes en adultos y ancianos, aunque son raros en niños y lactantes.4 Se han identificado en el periodo prenatal, aunque las lesiones prenatales suelen resolverse durante el embarazo.23
Epidemiología
La ecografía prenatal ha mostrado una prevalencia de quistes renales del 0,09%.23 La incidencia de quistes simples desde el nacimiento hasta los 18 años de edad tiene un promedio del 0,22% con un rango de 0,1% a 0,45%.24 La incidencia aumenta en los adultos y con la edad.
Patogenia
Existe mucha variedad en el tamaño; sin embargo, la mayoría miden menos de 2 cm y suelen ser corticales, alterando el contorno renal, pero también pueden ser corticales profundos o aparentemente medulares, aunque sin continuidad con la pelvis renal.1 Por lo general no afectan la función renal, pero en ocasiones pueden causar dolor cuando el quiste comprime estructuras adyacentes u obstruye el sistema colector.5
Evaluación y diagnóstico
La identificación suele ser incidental y el diagnóstico puede establecerse de forma segura mediante ecografía cuando se cumplen los siguientes criterios: 1) quiste internamente anecoico, 2) pared delgada, bien delimitada, con márgenes nítidos, 3) adecuada transmisión de las ondas de ultrasonido con refuerzo acústico posterior por detrás del quiste, 4) de forma esférica u ovoide.1 Si no se cumplen estos criterios o cuando la ecografía no proporciona una evaluación satisfactoria, como en el caso de quistes más complejos, la tomografía computarizada (TC) puede aportar mayor detalle anatómico. De forma alternativa, pueden estar indicadas la resonancia magnética (RM) o la aspiración con aguja.25
Los quistes simples no se comunican con la pelvis renal, sin embargo algunos quistes pueden aparecer en estrecha proximidad. La sospecha de comunicación puede evaluarse mediante imágenes seccionales con contraste intravenoso en fase tardía, como urografía por TC o urografía por RM. Además, la aspiración del quiste y las pruebas de laboratorio pueden ayudar a identificar la comunicación con la pelvis renal. Si no hay continuidad, el nitrógeno ureico en sangre y la creatinina serán comparables a los valores del paciente.5,26,27 Un quiste único también puede suscitar preocupación por ADPKD en el contexto de antecedentes familiares positivos.28 Históricamente, la clasificación de quistes de Bosniak no ha sido bien validada en niños, sin embargo un estudio reciente concluye que un sistema de clasificación de Bosniak modificado proporciona una estratificación de riesgo razonable y mostró que las lesiones con puntuación mBosniak 1 o 2 suelen ser benignas, mientras que las puntuaciones 3 o 4 presentaron patología intermedia o maligna en el 90% de los casos.29
Opciones de tratamiento y sus resultados
Los quistes sintomáticos pueden drenarse; sin embargo, es probable que recidiven si no se utiliza un agente esclerosante.5 Otra opción de manejo incluye la intervención quirúrgica con decorticación del quiste, que se presta a un abordaje mínimamente invasivo, laparoscópico, especialmente para quistes relativamente superficiales y/o exofíticos.
El seguimiento puede consistir en imágenes ecográficas para vigilar signos sugestivos de malignidad. Los quistes estables que no han crecido durante dos años pueden no requerir seguimiento adicional.29,30,31,32,33 La intervención puede no estar indicada una vez que se ha demostrado que la lesión no es maligna.
Divertículos caliciales
Los divertículos caliciales (CD) son saculaciones raras del cáliz que protruyen en el parénquima renal. Se clasifican en dos grupos: los de tipo 1, más frecuentes y adyacentes a un cáliz superior o inferior, mientras que los de tipo 2 son de mayor tamaño, se comunican con la pelvis renal y con mayor frecuencia son sintomáticos.34
Epidemiología
Se ha informado que el CD se encuentra en el 0,2% a 0,6% de los niños a quienes se les han realizado urogramas intravenosos.13
Patogénesis
La patogénesis de la CD se desconoce en gran medida y no se considera una enfermedad genéticamente hereditaria. Sin embargo, las teorías sí incluyen mutaciones génicas que inducen síndromes que afectan al riñón o, alternativamente, el fracaso de la regresión de la yema ureteral o causas obstructivas.13 El quiste está revestido por epitelio de células transicionales y se comunica con el sistema colector, típicamente a través de un cáliz o de un infundíbulo estrechado o estenosado y, con menor frecuencia, a través de la pelvis renal.35
Evaluación y diagnóstico
Los pacientes pueden estar asintomáticos o, por el contrario, presentarse con hematuria, dolor, ITU o nefrolitiasis; el signo de presentación más frecuente en niños es la ITU febril.35 Los divertículos asintomáticos suelen descubrirse en estudios de imagen. Aunque la ecografía puede identificar inicialmente el divertículo calicial, para distinguir entre un quiste simple y un divertículo calicial, el estudio de imagen más preciso es la imagen de corte transversal con contraste intravenoso y fase tardía.35,36,37 Figura 1 y Figura 2 demuestran hallazgos compatibles con DC en urografía por RM. Un estudio reciente que comparó la ecografía con la imagen de corte transversal encontró que la ecografía renal dirigida tuvo una sensibilidad del 40% y una especificidad del 100%, respectivamente, mientras que la imagen de corte transversal (específicamente la urografía por RM de líquido estático) tuvo una sensibilidad del 100% y una especificidad del 91,6%, respectivamente.37 El dolor, la infección, la formación de abscesos, la urosepsis y los cálculos renales sintomáticos pueden ser indicaciones posibles para el manejo quirúrgico.
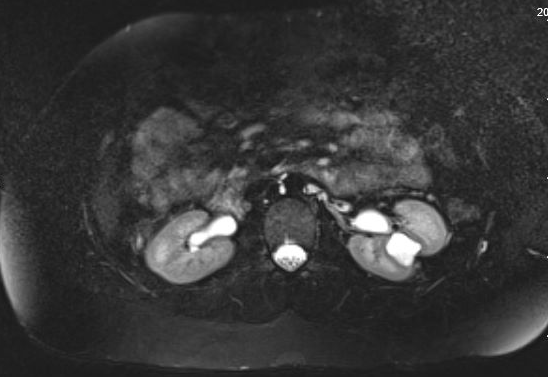
Figura 1 Urograma por RM que muestra opacificación por contraste de un divertículo calicial en la región interpolar del riñón izquierdo, con adelgazamiento suprayacente del parénquima renal.
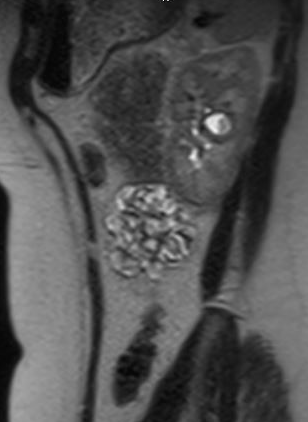
Figura 2 Vista sagital del urograma por RM en el mismo paciente, que muestra el contraste rellenando el divertículo calicial.
Opciones de tratamiento, sus resultados y complicaciones
Los pacientes asintomáticos no requieren tratamiento, pero pueden beneficiarse del seguimiento ultrasonográfico.1 Aquellos con indicaciones para manejo quirúrgico, como se mencionó anteriormente, pueden beneficiarse de la marsupialización laparoscópica del divertículo con fulguración del revestimiento epitelial.38,39,40 Otros procedimientos de manejo posibles pueden incluir ureteroscopia o nefrolitotomía percutánea mínimamente invasiva (en el contexto de cálculos),41 ablación ureteroscópica de la cavidad,39 o ablación y fulguración percutáneas.42 A pesar del abanico de posibilidades terapéuticas, la intervención quirúrgica para manejar el CD puede ser desafiante. Con el objetivo de minimizar la morbilidad, el manejo suele iniciarse con las opciones menos invasivas, como los abordajes endoscópicos o percutáneos. Puede haber una alta tasa de recurrencia de los síntomas,42 en cuyo caso la intervención puede escalar a abordajes laparoscópicos/robóticos e incluso abiertos, hasta incluir nefrectomía parcial o nefrectomía total. Por lo tanto, es necesario un asesoramiento exhaustivo a los pacientes al considerar la intervención quirúrgica para el CD.
Riñón multiquístico displásico
El riñón displásico multiquístico (MCDK) es una malformación congénita, diagnosticada con frecuencia durante la infancia o mediante ultrasonografía prenatal, caracterizada por numerosos quistes unilaterales no comunicantes y diferenciación metanéfrica anormal (presencia de cartílago, mesénquima indiferenciado y conductillos colectores inmaduros), a menudo en el contexto de una uropatía obstructiva.4,13,43 La enfermedad bilateral suele ser incompatible con la vida debido a oligohidramnios o anhidramnios, que conducen a hipoplasia pulmonar.44
Epidemiología
MCDK es una de las anomalías más frecuentes, con una incidencia estimada de 1 por cada 1.000 a 4.000 nacimientos.1,45 Es probable que una mayor incidencia de riñón único en la edad adulta esté asociada con la involución del MCDK a lo largo del tiempo, dada la baja incidencia de agenesia renal verdadera.5
Patogénesis
La displasia multiquística surge antes de la formación del nefrón, a partir de una diferenciación metanéfrica anómala, una alteración del blastema nefrogénico o una obstrucción de alto grado que ocurre durante el desarrollo renal.1 Los quistes están revestidos por un epitelio cúbico bajo, rodeado por células fusiformes, y pueden estar llenos de líquido proteináceo o hemático.1 Pueden observarse componentes primitivos y displásicos, así como cartílago inmaduro o glomérulos inmaduros. MCDK se clasifica como tipo infundíbulopélvico con atresia de la pelvis renal y del uréter, frente al tipo hidronefrótico, menos frecuente, que solo implica atresia de la porción superior del uréter.46 El riñón contralateral puede mostrar hipertrofia compensatoria.
Evaluación y diagnóstico
Actualmente, el MCDK se descubre con mayor frecuencia en la ecografía prenatal, mientras que la evaluación de una masa palpable en un recién nacido es la siguiente presentación más frecuente.47
Un diagnóstico de MCDK debe diferenciarse de la hidronefrosis debida a otras afecciones como la obstrucción de la unión pieloureteral (UPJ). Cabe destacar que, en MCDK, los quistes no se comunican y los quistes más grandes aparecen lateralmente. En la hidronefrosis, el parénquima renal rodea la estructura quística central y se mantiene la típica forma reniforme.48 Figura 3 y Figura 4 ayudan a demostrar las dificultades de diferenciar MCDK de la hidronefrosis en la ecografía. Figura 5 ilustra quistes no comunicantes de varios tamaños, típicos de MCDK. Para aclararlo aún más, puede realizarse una gammagrafía con radionúclidos que mostraría captación en la hidronefrosis y, por lo general, captación mínima o nula en el contexto de MCDK.49
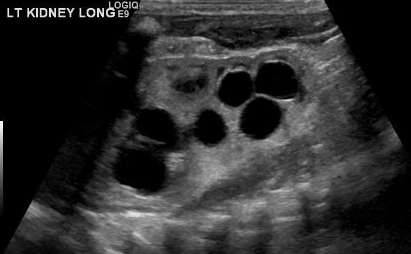
Figura 3 Ecografía renal que demuestra lo que podrían ser varios quistes renales en un MCDK frente a pielocaliectasia/hidronefrosis (HDN).
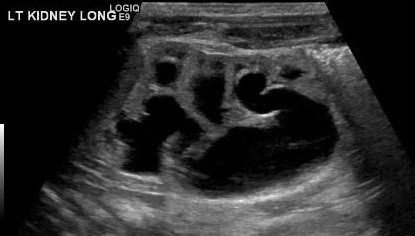
Figura 4 Otra imagen de ecografía renal de este mismo paciente, que demuestra continuidad entre la pelvis renal y los cálices, compatible con pielocaliectasia/HDN.
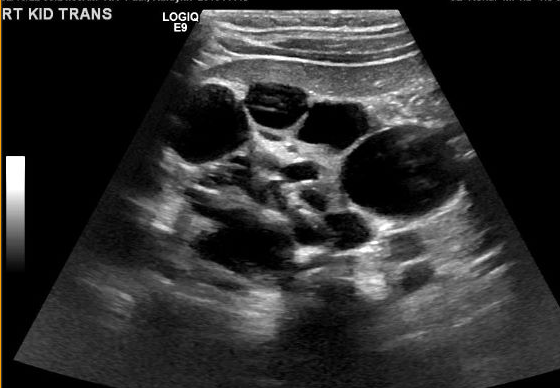
Figura 5 Ecografía renal que muestra múltiples quistes no comunicantes de diferentes tamaños, compatible con MCDK.
Opciones de tratamiento, sus resultados y complicaciones
La historia natural del MCDK implica involución con el tiempo, con posibles complicaciones que incluyen hipertensión y malignidad.5 Sin embargo, el riesgo de esta última puede ser bajo.50 Históricamente, los riñones afectados se extirpaban quirúrgicamente por la preocupación de los raros casos de degeneración maligna, pero ha habido un cambio hacia un manejo no operatorio, con ecografía de seguimiento de ambos riñones. Las indicaciones para cirugía incluyen cambios sospechosos de desarrollo de tumor de Wilms, efecto de masa, dolor, hipertensión y preferencia de los padres.5 A los pacientes con MCDK y un riñón contralateral normal se les debe tranquilizar respecto a la expectativa de una función renal normal a largo plazo, salvo que su riñón contralateral sufra una lesión. También se les debe aconsejar sobre la importancia de medidas de mantenimiento preventivo, como usar equipo de protección al practicar deportes, y adoptar hábitos de vida saludables (dieta y ejercicio) para mitigar otras causas de enfermedad renal (como la hipertensión y la diabetes). Para caracterizar el riesgo de desarrollo de enfermedad renal crónica, la vigilancia puede incluir además estudios de imagen del riñón contralateral, cistouretrografía miccional para cribado de RVU, control de la presión arterial y análisis de orina para detectar proteinuria.51
Riñón en esponja medular
El riñón en esponja medular (MSK) se caracteriza por dilatación de los conductos colectores distales, quistes y divertículos, todos restringidos a las pirámides medulares.1
Epidemiología
La MSK se observa principalmente en la adultez temprana, pero también puede presentarse en niños.52 Como muchas personas con MSK son asintomáticas, la incidencia no está claramente definida. Es más frecuente en personas con antecedentes de cálculos de calcio y se ha asociado con síndromes congénitos, como el síndrome de Beckwith-Wiedemann, el síndrome de Ehlers-Danlos, anodoncia y la enfermedad de Caroli.1
Patogénesis
MSK se considera en gran medida no hereditaria; sin embargo, ha habido casos que parecen heredarse en un patrón autosómico dominante, lo que respalda la hipótesis de que MSK interrumpe la interfaz entre la yema ureteral y el blastema metanéfrico.52
Los conductos colectores intrapapilares dilatados y los quistes medulares contienen material calcificado o descamado, confieren al riñón un aspecto esponjoso y son bilaterales en el 70% de los casos.1 Los quistes son contiguos a los túbulos colectores y están revestidos por epitelio del conducto colector.53
Evaluación y diagnóstico
El diagnóstico puede basarse en características urográficas de riñones aumentados de tamaño con calcificación ocasional, especialmente en las papilas, túbulos papilares dilatados que se llenan de contraste y realce de contraste papilar con opacificación medular residual.54 La evaluación hepática puede ayudar a diferenciar entre MSK y ADPKD si el diagnóstico no está claro.1
Opciones de tratamiento, sus resultados y complicaciones
El manejo está determinado principalmente por el tratamiento de los cálculos o de las infecciones. Se siguen medidas de prevención de cálculos, que incluyen aumento de la ingesta de líquidos, dieta baja en sodio, tiazidas y citrato de potasio.52 Las complicaciones asociadas con el MSK se relacionan con la mayor incidencia de nefrolitiasis y su morbilidad asociada.
Nefronoftisis
La nefronoftisis (NPHP) es una causa frecuente de enfermedad renal crónica en niños que se hereda de forma recesiva, a menudo se agrupa con la enfermedad renal quística medular (MCKD) porque comparten una apariencia radiográfica e histológica similar; sin embargo, la MCKD se hereda de manera autosómica dominante, presenta una manifestación tardía (edad adulta) y carece de afectación extrarrenal.5
Epidemiología
La NPHP se clasifica como infantil (1 año de edad), juvenil (13 años de edad) o adolescente (19 años de edad) según el defecto genético presente. La NPHP infantil conduce a IRT a los 1–3 años de edad y se asocia con mutaciones en NPHP2 (también denominado NPHP tipo 2). La NPHP juvenil, la manifestación más común, conduce a IRT a los 10–13 años y se asocia con mayor frecuencia con la mutación en NPHP1 (también denominado NPHP tipo 1). Por último, la NPHP adolescente se asocia con mutaciones en NPHP3 (también denominado NPHP tipo 3).5,55
Patogénesis
Aunque puede presentarse de forma esporádica, la NPHP suele heredarse con un patrón autosómico recesivo, siendo las mutaciones patogénicas homocigotas de NPHP1 en el cromosoma 2 la causa más frecuente.56 Existen muchos genes que se sabe están asociados con la NPHP (véase (Tabla 2)5
Tabla 2 Heterogeneidad genética de NPHP.5
| Tipo | Localización cromosómica | Gen mutado |
|---|---|---|
| NPH tipo 1 | 2q13 | NPHP1 |
| NPH tipo 2 | 9q22 | NPHP2 |
| NPH tipo 3 | 3q22 | NPHP3 |
| NPH tipo 4 | 1p36 | NPHP4 |
| NPH tipo 5 | 3q21 | NPHP5 |
| PH tipo 6 | 12q21 | NPHP6 |
| NPH tipo 7 | 16p | NPHP7 |
| NPH tipo 8 | 16q | NPHP8 |
| NPH tipo 9 | 17q11 | NPHP9 |
| NPH tipo 10 | 1q44 | SDCCAG8 |
| PH tipo 11 | 8q22 | MKS3 |
| AHI1 | 6q23 | AHI1 |
La evaluación macroscópica de los riñones con NPHP puede variar según el tipo, pero tienden a ser de pequeño tamaño y presentan una superficie granular. Histológicamente, muestran fibrosis intersticial e infiltración de células mononucleares, cambios tubulares variables que incluyen atrofia, engrosamiento y laminación de las membranas basales.57 Los quistes en la NPHP surgen de los túbulos contorneados distales y de los conductos colectores.57
Evaluación, diagnóstico y complicaciones
Los problemas con la capacidad de concentrar la orina pueden provocar poliuria y polidipsia (deshidratación, nicturia) con la consiguiente pérdida renal de sodio; síntomas de ERC como fatiga, anorexia y retraso del crecimiento; así como síntomas de osteodistrofia renal. La anemia puede estar presente de manera desproporcionada con respecto a la insuficiencia renal. La HTA es menos común excepto en la NPHP tipo 2.5,58 La afectación extrarrenal incluye fibrosis hepática y portal con hepatomegalia, además de muchos otros: síndrome de Senior-Løken (NPHP y degeneración tapetorretiniana), anomalías esqueléticas, discapacidad intelectual, ataxia cerebelosa, situs inversus, malformación cardíaca u otros síndromes menos frecuentes.5
La ecografía es un método común para la detección, pero puede no detectar todos los casos; la tomografía computarizada (TC) de cortes finos puede detectar dichos casos.58 Las pruebas genéticas están disponibles para el diagnóstico, al igual que la histología renal cuando las pruebas de imagen son inconcluyentes. Se recomienda realizar cribado hepático y ocular para la fibrosis hepática y el síndrome de Senior-Løken.5
Opciones de tratamiento y sus resultados
Hay pocos tratamientos eficaces disponibles para la NPHP. El manejo actual incluye tratamiento de soporte y medidas preventivas para la enfermedad renal crónica. Se prefiere el trasplante en el contexto de insuficiencia renal, ya que la lesión tubular no se produce en el riñón del donante.13
Quistes multiloculares
El quiste multilocular (nefroma quístico) es una proliferación neoplásica benigna que se presenta en adultos y niños y forma parte de un espectro con el tumor de Wilms.
Epidemiología
La mayoría de los casos ocurren antes de los 4 años de edad o después de los 30 años de edad, con un predominio masculino en la población pediátrica.59
Patogénesis
Las lesiones son voluminosas, no comunicantes, bien encapsuladas por tejido fibroso, pueden contener tejido embrionario, y pueden comprimir el parénquima renal normal.5
Evaluación, diagnóstico y opciones de tratamiento
Los quistes multiloculares en niños generalmente se presentan con una masa abdominal palpable.60 La evaluación se realiza con ecografía o tomografía computarizada, sin embargo, debido a la dificultad que existe en ocasiones para diferenciar entre los quistes multiloculares y el tumor de Wilms quístico basándose solo en los estudios de imagen, el diagnóstico definitivo puede establecerse mediante examen histopatológico después de una nefrectomía o una nefrectomía parcial.60
Enfermedades quísticas renales adquiridas
La enfermedad renal quística adquirida (ACKD) describe los cambios quísticos bilaterales de los riñones en el contexto de ESRD y azoemia y no enfermedad renal quística hereditaria.1
Epidemiología
Se observan quistes en el 10% de los pacientes con ERT, aumentando hasta el 44% y el 60% a los 3 y 5 años tras el inicio de la diálisis.1
Patogénesis
Se considera que la etiología está mediada por toxinas, la acumulación de factores de crecimiento, o la obstrucción tubular debido a fibrosis, cristales de oxalato, oclusión vascular o isquemia.1 Los riñones están afectados bilateralmente y suelen ser de menor tamaño que lo normal, mientras que la histología muestra un epitelio plano a cuboidal similar al epitelio tubular distal.4
Evaluación, diagnóstico, tratamiento y complicaciones
La mayoría de los pacientes con quistes están asintomáticos, pero pueden presentarse con dolor y/o hematuria debido a sangrado espontáneo dentro de los quistes. La ecografía suele utilizarse para el diagnóstico y el seguimiento, mientras que la TC o la RM pueden identificar los quistes con mayor facilidad; sin embargo, en pacientes con insuficiencia renal terminal (IRT) la obtención de imágenes con contraste intravenoso debe manejarse cuidadosamente debido a los efectos nefrotóxicos de los agentes de contraste.1 Las principales complicaciones son la hemorragia y la transformación neoplásica.4 Se recomienda ecografía de vigilancia, y nefrectomía en un contexto sospechoso de neoplasia.5 Por lo tanto, el tratamiento se maneja de forma conservadora con control de los síntomas y ajuste de los calendarios de cribado o de los regímenes de diálisis.
Síndromes asociados con la enfermedad renal quística
Los quistes renales pueden observarse en diversos síndromes, entre los más comunes se incluyen la esclerosis tuberosa autosómica dominante y el síndrome de Von Hippel-Lindau. Otros con patrones de herencia autosómica recesiva incluyen el síndrome de Meckel, la distrofia torácica asfixiante de Jeune y el síndrome cerebrohepatorenal de Zellweger.1
Esclerosis tuberosa
La esclerosis tuberosa se diagnostica por la presencia de dos criterios mayores o de uno mayor más dos criterios menores (véase Tabla 3).61 Los quistes renales múltiples se incluyen como criterio menor.61,62 La afectación renal es una causa importante de morbilidad y mortalidad en la esclerosis tuberosa.63
Tabla 3 Criterios mayores y menores para el complejo de esclerosis tuberosa.61
| Criterios mayores | Criterios menores |
|---|---|
| Máculas hipomelanóticas (≥3, de al menos 5 mm de diámetro) | Lesiones cutáneas en “confeti” |
| Angiofibromas (≥3) o placa fibrosa cefálica | Fosetas del esmalte dental (>3) |
| Fibromas ungueales (≥2) | Fibromas intraorales (≥2) |
| Placa de Shagreen | Parche acrómico retiniano |
| Hamartomas retinianos múltiples | Quistes renales múltiples |
| Displasias corticales | Hamartomas no renales |
| Nódulos subependimarios | |
| Astrocitoma subependimario de células gigantes | |
| Rabdomioma cardíaco | |
| Linfangioleiomiomatosis (LAM) | |
| Angiomiolipomas (≥2) |
Síndrome de Von Hippel-Lindau
El síndrome de Von Hippel-Lindau, de manera similar, tiene numerosas manifestaciones, entre las cuales el carcinoma de células renales y los quistes renales son comunes, pero rara vez se observan en niños.64
Síndrome de Meckel
Esto puede presentarse con una variedad de manifestaciones, incluyendo quistes. La displasia quística siempre está presente.5,65
Conclusiones
Los quistes renales llenos de líquido se encuentran en un grupo amplio y heterogéneo de enfermedades quísticas renales. Aunque comparten una manifestación común en forma de quistes renales, este grupo de enfermedades se diferencia por muchos factores: patrón de herencia, edad de inicio, anomalías renales o extrarrenales asociadas, curso clínico, diferencias histológicas, aspecto en las pruebas de imagen, entre otros. La identificación precisa de un proceso patológico puede ser difícil cuando hay superposición de la sintomatología. Las técnicas de imagen, especialmente la ecografía, han sido durante mucho tiempo un modo principal de identificar muchas de estas enfermedades. Las opciones de pruebas genéticas son cada vez más comunes y accesibles.
La investigación continua conduce a avances en la ciencia básica y en la clínica, ayudando a identificar nuevas enfermedades, fisiopatología, modalidades diagnósticas y opciones de tratamiento.
Puntos clave
- La distinción entre un quiste simple y un divertículo calicial puede resultar difícil; sin embargo, es de especial importancia, ya que un divertículo calicial tiene mucha más probabilidad de causar síntomas y requerir intervención. El diagnóstico puede facilitarse mediante imágenes de sección transversal con contraste IV y en fase tardía.
- También es importante distinguir entre MCDK y la hidronefrosis severa, como en el contexto de una obstrucción de la unión ureteropélvica (UPJ). El manejo de MCDK es en gran medida observacional, mientras que existe una mayor probabilidad de que la intervención sea necesaria en la hidronefrosis o la obstrucción de la UPJ. El diagnóstico puede facilitarse mediante imágenes con radionúclidos.
- El manejo de ciertas enfermedades renales quísticas, como ADPKD y ARPKD, debe realizarse en colaboración con nefrología para facilitar una atención médica renal óptima.
- La caracterización de los quistes mediante imágenes prenatales a menudo ayuda a diferenciar entre posibles enfermedades (véase la Figura 6 más abajo).13

Figura 6 Diferenciación de quistes mediante imágenes prenatales.13
Lecturas sugeridas
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
Referencias
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- Glassberg KI, Stephens FD, Lebowitz RL. Renal dysgenesis and cystic disease of the kidney: a report of the Committee on Terminology, Nomenclature and Classification, Section on Urology, American Academy of Pediatrics. J Urol Oct 1987; 138 (4 Pt 2): 1085–1092. DOI: 10.1016/s0022-5347(17)43510-5.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364 (16): 1533–1543. DOI: 10.1056/NEJMra1010172.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Cole BR, Conley SB, Stapleton FB. Polycystic kidney disease in the first year of life. J Pediatr Nov 1987; 111 (5): 693–699. DOI: 10.1016/s0022-3476(87)80244-5.
- Gabow PA. Polycystic kidney disease: clues to pathogenesis. Kidney Int Dec 1991; 40 (6): 989–996. DOI: 10.1038/ki.1991.306.
- Proesmans W, Damme B, Casaer P, Marchal G. Autosomal dominant polycystic kidney disease in the neonatal period: association with a cerebral arteriovenous malformation. Pediatrics Dec 1982; 70 (6): 971–975. DOI: 10.1542/peds.70.6.971.
- Ong AC, Wheatley DN. Polycystic kidney disease–the ciliary connection. Lancet Mar 2003; 361 (9359): 774–776. DOI: 10.1016/S0140-6736(03)12662-1.
- Rossetti S, Consugar MB, Chapman AB. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol Jul 2007; 18 (7): 2143–2160. DOI: 10.1681/ASN.2006121387.
- Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol Oct 2004; 15 (10): 2528–2536. DOI: 10.1097/01.ASN.0000141055.57643.E0.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet Apr 1994; 343 (8901): 824–827. DOI: 10.1016/s0140-6736(94)92026-5.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Grantham JJ. Polycystic kidney disease: hereditary and acquired. Adv Intern Med 1993; 38: 409–420.
- Schrier R, McFann K, Johnson A. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol Jul 2002; 13 (7): 1733–1739. DOI: 10.1097/01.asn.0000018407.60002.b9.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med Oct 1990; 323 (16): 1091–1096. DOI: 10.1056/NEJM199010183231602.
- Alzarka B, Morizono H, Bollman JW, Kim D, Guay-Woodford LM. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Front Pediatr 2017; 5 (80). DOI: 10.3389/fped.2017.00080.
- Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers 2018; 4 (1). DOI: 10.1038/s41572-018-0047-y.
- Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatr Nephrol Jan 1989; 3 (1): 43–49. DOI: 10.1007/BF00859625.
- Ward CJ, Hogan MC, Rossetti S. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet Mar 2002; 30 (3): 259–269. DOI: 10.1038/ng833.
- Onuchic LF, Furu L, Nagasawa Y. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet May 2002; 70 (5): 1305–1317. DOI: 10.1086/340448.
- Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol Jun 1997; 11 (3): 302–306. DOI: 10.1007/s004670050281.
- Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics Sep 2014; 134 (3). DOI: 10.1542/peds.2013-3646.
- Blazer S, Zimmer EZ, Blumenfeld Z, Zelikovic I, Bronshtein M. Natural history of fetal simple renal cysts detected in early pregnancy. J Urol 1999; 162 (3 Pt 1): 812–814. DOI: 10.1097/00005392-199909010-00066.
- McHugh K, Stringer DA, Hebert D, Babiak CA. Simple renal cysts in children: diagnosis and follow-up with US. Radiology Feb 1991; 178 (2): 383–385. DOI: 10.1148/radiology.178.2.1987597.
- Bosniak MA. The current radiological approach to renal cysts. Radiology Jan 1986; 158 (1): 1–10. DOI: 10.1148/radiology.158.1.3510019.
- Lee J, Darcy M. Renal cysts and urinomas. Semin Intervent Radiol. Dec 2011; 28 (4): 380–391. DOI: 10.1055/s-0031-1296080.
- Steinhardt GF, Slovis TL, Perlmutter AD. Simple renal cysts in infants. Radiology May 1985; 155 (2): 349–350. DOI: 10.1148/radiology.155.2.3885305.
- Gabow PA, Kimberling WJ, Strain JD, Manco-Johnson ML, Johnson AM. Utility of ultrasonography in the diagnosis of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol Jan 1997; 8 (1): 105–110. DOI: 10.1681/ASN.V81105.
- Peard L, Gargollo P, Grant C. Validation of the modified Bosniak classification system to risk stratify pediatric cystic renal masses: An international, multi-site study from the pediatric urologic oncology working group of the societies for pediatric urology. J Pediatr Urol 2022; 18 (2). DOI: 10.1016/j.jpurol.2021.12.001.
- Bayram MT, Alaygut D, Soylu A, Serdaroğlu E, Cakmakçı H, Kavukçu S. Clinical and radiological course of simple renal cysts in children. Urology Feb 2014; 83 (2): 433–437. DOI: 10.1016/j.urology.2013.08.055.
- Karmazyn B, Tawadros A, Delaney LR. Ultrasound classification of solitary renal cysts in children. J Pediatr Urol Jun 2015; 11 (3). DOI: 10.1016/j.jpurol.2015.03.001.
- O’Kelly F, McAlpine K, Abdeen N, Keays MA, Leonard MP, Guerra LA. The Prevalence, Clinicodemographics, and Outcomes of Incidental and Symptomatic Renal Cysts in a Pediatric Cohort Undergoing Ultrasonography. J Urol 2019; 202 (2): 394–399. DOI: 10.1097/JU.0000000000000264.
- Rediger C, Guerra LA, Keays MA. Renal cyst evolution in childhood: a contemporary observational study. J Pediatr Urol Apr 2019; 15 (2). DOI: 10.1016/j.jpurol.2019.01.006.
- Wulfsohn MA. Pyelocaliceal diverticula. J Urol Jan 1980; 123 (1): 1–8. DOI: 10.1016/s0022-5347(17)55748-1.
- Estrada CR, Datta S, Schneck FX, Bauer SB, Peters CA, Retik AB. Caliceal diverticula in children: natural history and management. J Urol Mar 2009; 181 (3): 1311. DOI: 10.1016/j.juro.2008.10.043.
- Waingankar N, Hayek S, Smith AD, Okeke Z. Calyceal diverticula: a comprehensive review. Rev Urol 2014; 16 (1): 29–43.
- Sahin H, Sarioglu FC, Alaygut D, Akdogan AI, Pekcevik Y. Differentiation of simple renal parenchymal cyst and calyceal diverticulum. Pediatr Int May 2020; 62 (5): 615–623. DOI: 10.1111/ped.14127.
- Casale P, Grady RW, Feng WC, Joyner BD, Mitchell ME. The pediatric caliceal diverticulum: diagnosis and laparoscopic management. J Endourol Sep 2004; 18 (7): 668–671. DOI: 10.1089/end.2004.18.668.
- Long CJ, Weiss DA, Kolon TF, Srinivasan AK, Shukla AR. Pediatric calyceal diverticulum treatment: An experience with endoscopic and laparoscopic approaches. J Pediatr Urol Aug 2015; 11 (4). DOI: 10.1016/j.jpurol.2015.04.013.
- Sripathi V, Mitra A, Padankatti RL, Ganesan T. Robotic treatment of a type 2 calyceal diverticulum in a child: is suture closure and marsupialisation enough for a good outcome? J Robot Surg Dec 2018; 12 (4): 727–730. DOI: 10.1007/s11701-017-0758-1.
- Ding X, Xu ST, Huang YH. Management of symptomatic caliceal diverticular calculi: Minimally invasive percutaneous nephrolithotomy versus flexible ureterorenoscopy. Chronic Dis Transl Med Dec 2016; 2 (4): 250–256. DOI: 10.1016/j.cdtm.2016.11.016.
- Monga M, Smith R, Ferral H, Thomas R. Percutaneous ablation of caliceal diverticulum: long-term followup. J Urol Jan 2000; 163 (1): 28–32. DOI: 10.1016/s0022-5347(05)67965-7.
- Gilbert-Barness E, Potter EL. Respiratory system. 2nd ed., DOI: 10.1001/jama.1997.03540320078045.
- D’Alton M, Romero R, Grannum P, DePalma L, Jeanty P, Hobbins JC. Antenatal diagnosis of renal anomalies with ultrasound. IV Bilateral Multicystic Kidney Disease Am J Obstet Gynecol Mar 1986; 154 (3): 532–537. DOI: 10.1016/0002-9378(86)90597-1.
- Kalyoussef E, Hwang J, Prasad V, Barone J. Segmental multicystic dysplastic kidney in children. Urology Nov 2006; 68 (5). DOI: 10.1016/j.urology.2006.06.024.
- Felson B, Cussen LJ. The hydronephrotic type of unilateral congenital multicystic disease of the kidney. Semin Roentgenol Apr 1975; 10 (2): 113–123. DOI: 10.1016/0037-198x(75)90035-8.
- Welch TR, Wacksman J. The changing approach to multicystic dysplastic kidney in children. J Pediatr Jun 2005; 146 (6): 723–725. DOI: 10.1016/j.jpeds.2005.02.027.
- Sanders RC, Hartman DS. The sonographic distinction between neonatal multicystic kidney and hydronephrosis. Radiology Jun 1984; 151 (3): 621–625. DOI: 10.1148/radiology.151.3.6718720.
- Roach PJ, Paltiel HJ, Perez-Atayde A, Tello RJ, Davis RT, Treves ST. Renal dysplasia in infants: appearance on 99mTc DMSA scintigraphy. Pediatr Radiol 1995; 25 (6): 472–475. DOI: 10.1007/BF02019071.
- Narchi H. Risk of Wilms’ tumour with multicystic kidney disease: a systematic review. Arch Dis Child Feb 2005; 90 (2): 147–149. DOI: 10.1136/adc.2004.051243.
- Mansoor O, Chandar J, Rodriguez MM. Long-term risk of chronic kidney disease in unilateral multicystic dysplastic kidney. Pediatr Nephrol Apr 2011; 26 (4): 597–603. DOI: 10.1007/s00467-010-1746-0.
- Gambaro G, Danza FM, Fabris A. Medullary sponge kidney. Curr Opin Nephrol Hypertens. Jul 2013; 22 (4): 421–426. DOI: 10.1097/MNH.0b013e3283622b86.
- Bernstein J. The classification of renal cysts. Nephron 1973; 11 (2): 91–100. DOI: 10.1159/000180222.
- Gedroyc WM, Saxton HM. More medullary sponge variants. Clin Radiol Jul 1988; 39 (4): 423–425. DOI: 10.1016/s0009-9260(88)80292-7.
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol Jan 2009; 20 (1): 23–35. DOI: 10.1681/ASN.2008050456.
- Hildebrandt F, Otto E, Rensing C. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet Oct 1997; 17 (2): 149–153. DOI: 10.1038/ng1097-149.
- Sherman FE, Studnicki FM, Fetterman G. Renal lesions of familial juvenile nephronophthisis examined by microdissection. Am J Clin Pathol Apr 1971; 55 (4): 391–400. DOI: 10.1093/ajcp/55.4.391.
- Elzouki AY, al-Suhaibani H, Mirza K, al-Sowailem AM. Thin-section computed tomography scans detect medullary cysts in patients believed to have juvenile nephronophthisis. Am J Kidney Dis Feb 1996; 27 (2): 216–219. DOI: 10.1016/s0272-6386(96)90543-0.
- Eble JN, Bonsib SM. Extensively cystic renal neoplasms: cystic nephroma, cystic partially differentiated nephroblastoma, multilocular cystic renal cell carcinoma, and cystic hamartoma of renal pelvis. Semin Diagn Pathol Feb 1998; 15 (1): 2–20.
- Castillo OA, Boyle ET, Kramer SA. Multilocular cysts of kidney. A study of 29 patients and review of literature. Urology Feb 1991; 37 (2): 156–162. DOI: 10.1016/0090-4295(91)80214-r.
- Northrup H, Krueger DA, ITSCC G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol Oct 2013; 49 (4): 243–254. DOI: 10.1016/j.pediatrneurol.2013.08.001.
- Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol Dec 1998; 13 (12): 624–628. DOI: 10.1177/088307389801301206.
- Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc Aug 1991; 66 (8): 792–796. DOI: 10.1016/s0025-6196(12)61196-3.
- Maddock IR, Moran A, Maher ER. A genetic register for von Hippel-Lindau disease. J Med Genet Feb 1996; 33 (2): 120–127. DOI: 10.1136/jmg.33.2.120.
- Salonen R. The Meckel syndrome: clinicopathological findings in 67 patients. Am J Med Genet Aug 1984; 18 (4): 671–689. DOI: 10.1002/ajmg.1320180414.
Última actualización: 2025-09-21 13:35