
13: Cystic Diseases of the Kidney
This chapter will take approximately 21 minutes to read.
Introduction
Renal cystic diseases make up a broad category of sporadic and genetic, congenital or acquired renal conditions that may involve the presence of a single cyst to many cysts of one or both kidneys (see heritable classifications in Table 1 below).1 Based on a 1987 Committee on Classification, Nomenclature, and Terminology, classification of renal cystic disease is primarily determined by genetic and nongenetic disease.2 Renal cystic diseases may largely be considered rare conditions, however they may be associated with serious consequences, such as chronic kidney disease (CKD), kidney failure, or hepatic disease. Despite their rarity, autosomal dominant polycystic kidney disease (ADPKD) is one of the most common inherited human conditions. The possibility of serious complications from renal cystic diseases in the pediatric population, especially CKD, make early diagnosis and effective treatment modalities of certain importance.
Table 1 Heritable classifications of renal cystic diseases.1
| Inheritable | Nonheritable |
|---|---|
| Autosomal recessive (infantile) polycystic kidney disease | Multicystic kidney (multicystic dysplastic kidney) |
| Autosomal dominant (adult) polycystic kidney disease | Benign multilocular cyst (cystic nephroma) |
| Juvenile nephronophthisis and medullary cystic disease complex | Simple cysts |
| Juvenile nephronophthisis (autosomal recessive) | Medullary sponge kidney |
| Medullary cystic disease (autosomal dominant) | Sporadic glomerulocystic kidney disease |
| Congenital nephrosis (familial nephrotic syndrome) (autosomal recessive) | Acquired renal cystic disease |
| Familial hypoplastic glomerulocystic disease (autosomal dominant) | Calyceal diverticulum (pyelogenic cyst) |
| Multiple malformation syndromes with renal cysts (e.g., tuberous sclerosis, von Hippel-Lindau disease) |
Embryology
Cysts typically occur secondary to secretory, absorptive, and electrolyte imbalances across epithelial cells and form anywhere within the renal tubular system. Recent research for inherited cystic renal disease suggests affected genes involve primary cilium of renal epithelial cells, a potential common pathway for these conditions.3 The term “ciliopathy” refers to this common pathway. The cilia function to modulate proliferation and differentiation of tubular cells, and their dysfunction may lead to inappropriate tubule expansion and cyst formation.4
This chapter will discuss the epidemiology, pathogenesis, evaluation, and treatment of various heritable and nonheritable cystic renal diseases, including ADPKD, autosomal recessive polycystic renal disease (ARPKD), isolated renal cysts, calyceal diverticuli, multicystic dysplastic kidney (MCDK), medullary sponge kidney, nephronophthisis, multilocular cysts (multilocular cystic nephroma), acquired cystic kidney diseases, and syndromes associated with cystic kidney disease.
Autosomal Dominant Polycystic Kidney Disease
ADPKD is relatively common, with an incidence of 1 in 400 to 1,000 live births manifesting as cystic dilation of the nephron and other extrarenal abnormalities, such as intracranial aneurysm.5 Severity varies per patient and tends to worsen over time, thus many pediatric patients and even some middle-aged adults can be asymptomatic. Although uncommon, severe disease in childhood may portend significant morbidity and mortality.6 Due to family history or incidental imaging findings, more asymptomatic children are being identified with ADPKD.
Epidemiology
Of those affected with ADPKD, about 96% will have clinical signs of the disease by the age of 90.7 Despite the majority of cases being discovered in middle-aged patients (30–40 years old) and rare renal failure before 40 years of age, it has been described as early as the newborn period. When described in newborns, ADPKD is felt to be more aggressive.8 Despite the autosomal dominant inheritance pattern with theoretical 100% penetrance, 10% of cases may occur sporadically.1
Pathogenesis
ADPKD is inherited in an autosomal dominant fashion and the major genetic mutations occur in PKD1 and PKD2 on chromosome 16 and chromosome 4, respectively.5 The gene product is a transmembrane protein (polycystin 1 and polycystin 2, respectively) localized within primary cilia.9 PKD1 mutations on chromosome 16 account for the majority of cases and increased severity while most remaining cases are from PKD2 mutations.10
Improper functioning of polycystin 1 or polycystin 2 leads to dysregulation of proliferative pathway signals such as cAMP, ERK, and mTOR. Cilia may play an organizational role in transduction of these signals, as dysregulation leads to cyst formation.11
Evaluation and Diagnosis
As a cost effective and minimally invasive imaging study, renal ultrasonography is frequently used for identifying ADPKD. Ultrasonographic diagnostic criteria in those younger than 30 include the presence of at least two unilateral or bilateral cysts, with more cysts needed to suggest diagnosis as age increases.12 However, ADPKD and ARPKD can be difficult to distinguish in the pediatric population, making the presence of positive family history crucial in a diagnosis.13 In the absence of family history, genetic testing can be performed. Other factors important to consider, when family history is negative, include bilateral renal enlargement, three or more hepatic cysts, cerebral artery aneurysm, and solitary cysts of other organs (arachnoid, pineal gland, pancreas, or spleen).1,14 Imaging or biopsy of the liver may also help differentiate ADPKD and ARPKD as hepatic fibrosis is always present in ARPKD, but rare in ADPKD.5 Finally, renal pathology (biopsy) may be helpful as ADPKD may involve the entirety of the tubule, including the glomerulus, however ARPKD would not have glomerular cysts.5
Treatment Options, Outcomes, and Complications
There is currently no cure for ADPKD. Goals of treatment include delaying onset of end-stage renal disease (ESRD) and reducing complication burden of ADPKD related to renal function decline, cardiac disease, and intracranial hemorrhage. Complications are most significantly modulated by managing hypertension.1 Therefore, rigorous control of blood pressure is recommended and angiotensin-converting enzyme (ACE) inhibitors and angiotensin-receptor blockers are a common choice, however there is no consensus on ideal antihypertensives.15,16
Emerging therapies under investigation aim to limit cyst growth by reducing cAMP using mTOR inhibitors, somatostatin, tyrosine kinase inhibitors, Src kinase inhibitors, MEK inhibitors, and cyclin-dependent kinase inhibitors.
Autosomal Recessive Polycystic Kidney Disease
ARPKD is characterized by rapid enlargement of both kidneys in infants due to the formation of cysts within the collecting duct system. It may be associated with congenital hepatic fibrosis which can lead to portal hypertension. An earlier presentation of ARPKD is often associated with increased severity.1 Its multisystem effects may necessitate multidisciplinary management.
Epidemiology
As ARPKD is less common than ADPKD, there is less epidemiological data. Reported incidence is 1 in 26,500 live births but may range as much as 1 in 10,000 to 50,000 live births.1,17 However, it may be more common in isolated or consanguineous populations.
Pathogenesis
ARPKD is inherited in a classically autosomal recessive pattern, with unaffected heterozygote parents having a 25% risk of having an affected child. It is caused by mutation of chromosome 6 in the polycystic kidney and hepatic disease 1 (PKHD1) gene which encodes fibrocystin (also known as polyductin), a protein identified in the renal tubule system (collecting duct and thick ascending limb), epithelial cells of hepatic bile duct, and in primary apical cilia.5,18,19
A typical patient may present in the neonatal period with a history of prenatal oligohydramnios, enlarged kidneys, and “Potter” sequence with pulmonary hypoplasia and perinatal death in approximately 30% of affected newborns.20,21 Enlarged kidneys appear echogenic with fusiform collecting duct dilation and progression to ESRD may occur at varying ages. Effect on other organ systems may manifest as dilated biliary ducts, congenital hepatic fibrosis, portal hypertension, and neurocognitive dysfunction.22
Evaluation and Diagnosis
Fetal and newborn ultrasound may show bilateral, very enlarged, and diffusely echogenic kidneys due to abundant microcysts.1 The presence of macrocysts (> 10 mm) may indicate other ciliopathies such as MCDK or ADPKD and MRI may help further evaluate anomalies in the setting of severe oligohydramnios.13 Accurate diagnosis depends on suggestive imaging findings, presence of family history with recessive mode of inheritance, liver biopsy showing periportal lesions (present in ARPKD and rare in ADPKD), and lack of extrarenal manifestations associated with other cystic renal diseases.1
Treatment Options, Outcomes, and Complications
Similar to ADPKD, there is no cure for ARPKD, thus many treatment options are for symptom management or palliation related to hypertension, congestive heart failure, and renal and hepatic failure. Aggressive treatment, such as unilateral or bilateral nephrectomy, may be indicated in severely affected individuals who suffer respiratory or nutritional compromise due to mass effect from enlarged kidneys. Decompressive therapies may be implemented for management of portal hypertension. Hemodialysis and renal transplantation are often considered.1
Isolated Renal Cysts
Solitary and multiple renal cysts are the most common lesions of the adult and elderly kidney, yet rare in children and infants.4 They have been identified prenatally, although prenatal lesions commonly resolve during pregnancy.23
Epidemiology
Prenatal ultrasound has shown prevalence of renal cysts to be 0.09%.23 Incidence of simple cysts from birth to 18 years of age averages at 0.22% with a range of 0.1% to 0.45%.24 Incidence rises in adults and advancing age.
Pathogenesis
There is much variety in size, however most remain less than 2 cm and are often cortical, disrupting renal contour, but may also be deep cortical or seemingly medullary, but without continuity with the renal pelvis.1 They typically do not impair renal function, but may occasionally cause pain when the cyst compresses adjacent structures or obstructs the collecting system.5
Evaluation and Diagnosis
Identification is typically incidental and diagnosis can be safely made by ultrasound when meeting the following criteria: 1) internally anechoic cyst, 2) thin, sharply-defined wall with distinct margins, 3) adequate transmission of ultrasound waves with enhancement behind cyst, 4) spherical or ovoid in shape.1 If these criteria are not met or when ultrasound does not provide satisfactory evaluation, as in the case of more complex cysts, CT scan may provide more anatomic detail. Alternatively, MRI or needle aspiration may be indicated.25
Simple cysts do not communicate with the renal pelvis, however some cysts may appear in close proximity. Concern for communication can be evaluated by cross-sectional imaging with IV contrast with delayed phase, such as CT urogram or MR urogram. Additionally, cyst aspiration and lab testing can help identify renal pelvis communication. If noncontiguous, blood urea nitrogen and creatinine will be comparable to the patients values.5,26,27 A single cyst may also raise concern for ADPKD in the setting of positive family history.28 Historically the Bosniak cyst classification has not been well validated in children, however one recent study concludes that a modified Bosniak classification system provides reasonable risk stratification and showed that lesions with mBosniak score 1 or 2 are often benign, while scores 3 or 4 proved to have 90% containing intermediate or malignant pathology.29
Treatment Options and Their Outcomes
Symptomatic cysts may be drained, however are likely to recur without the use of a sclerosing agent.5 Another management option includes surgical intervention with cyst decortication, which is amenable to a minimally invasive, laparoscopic approach, especially for cysts that are relatively superficial and/or exophytic.
Follow up may consist of ultrasonographic imaging to monitor for signs suggestive of malignancy. Stable cysts that have not grown for two years may require no additional follow-up.29,30,31,32,33 Intervention may not be indicated after lesion has been proven to not be malignant.
Calyceal Diverticuli
Calyceal diverticuli (CD) are rare outpouchings of calyx into the renal parenchyma. They are classified into two groups: type 1 being more common and adjacent to an upper or lower calyx while type 2 are larger, communicate with the renal pelvis, and are more often symptomatic.34
Epidemiology
CD has been reported to be found in 0.2% to 0.6% of children who have had IV urograms performed.13
Pathogenesis
The pathogenesis of CD is largely unknown and is not considered a genetically inherited disease. However, theories do include gene mutations inducing syndromes that affect the kidney, or alternatively failure of ureteric bud regression or obstructive causes.13 The cyst is lined with transitional cell epithelium and communicates with the collecting system, typically through a calyx or narrowed or stenosed infundibulum and less often through the renal pelvis.35
Evaluation and Diagnosis
Patients may be asymptomatic or rather present with hematuria, pain, UTI, or nephrolithiasis; the most common presenting sign in children is febrile UTI.35 Asymptomatic diverticula are typically discovered on imaging studies. While ultrasound may initially identify the calyceal diverticulum, in order to distinguish between a simple cyst and a calyceal diverticulum, the most accurate imaging study is cross-sectional imaging with IV contrast and a delayed phase.35,36,37 Figure 1 and Figure 2 demonstrate findings consistent with CD on MR urography. A recent study comparing ultrasound to cross-sectional imaging found that focused renal US had a sensitivity of 40% and specificity of 100% respectively, while cross-sectional imaging (specifically static-fluid MR urography) had a sensitivity of 100% and specificity of 91.6% respectively.37 Pain, infection, abscess formation, urosepsis, symptomatic renal calculi may all be possible indications for surgical management.
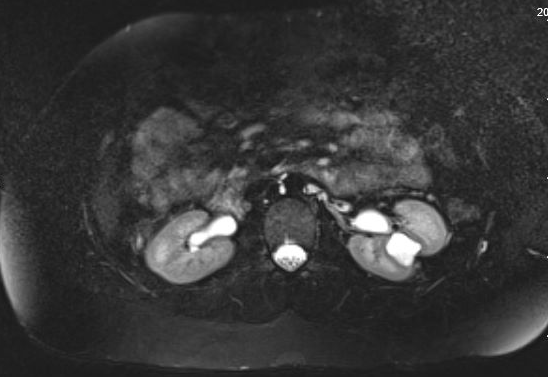
Figure 1 MR Urogram showing contrast filling a calyceal diverticulum in the interpolar region of the left kidney with overlying thinning of the renal parenchyma.
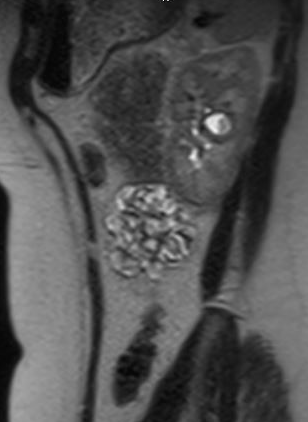
Figure 2 Sagittal view of MR urogram in same patient, showing contrast filling the calyceal diverticulum.
Treatment Options, Their Outcomes, and Complications
Asymptomatic patients do not require treatment, but may benefit from ultrasonographic monitoring.1 Those with indications for surgical management, as mentioned above, may benefit from laparoscopic marsupialization of diverticulum with fulguration of epithelial lining.38,39,40 Other possible management procedures may include ureteroscopy or minimally invasive percutaneous nephrolithotomy (in setting of stones),41 ureteroscopic ablation of cavity,39 or percutaneous ablation and fulguration.42 Despite the array of treatment possibilities, surgical intervention to manage CD can be challenging. In an attempt to minimize morbidity, management typically begins with the less invasive options, such as endoscopic or percutaneous approaches. There can be a high rate of symptom recurrence,42 in which case intervention can escalate to laparoscopic/robotic approaches and even open approaches, up to and including partial nephrectomy or total nephrectomy. Therefore, extensive counseling of patients is necessary when considering surgical intervention for CD.
Multicystic Dysplastic Kidney
Multicystic dysplastic kidney (MCDK) is a congenital malformation commonly diagnosed during childhood or by prenatal ultrasonography characterized by many unilateral noncommunicating cysts and abnormal metanephric differentiation (presence of cartilage, undifferentiated mesenchyme, and immature collecting ductules) often in setting of obstructive uropathy.4,13,43 Bilateral disease is usually incompatible with life due to oligohydramnios or anhydramnios leading to pulmonary hypoplasia.44
Epidemiology
MCDK is one of the most common anomalies, with an estimated incidence of 1 in 1,000 to 4,000 births.1,45 A higher incidence of solitary kidney in adulthood is likely associated with involution of MCDK over time given the low incidence of true renal agenesis.5
Pathogenesis
Multicystic dysplasia arises before formation of the nephron, from abnormal metanephric differentiation, an abnormality of the nephrogenic blastema, or high-grade obstruction occurring during kidney development.1 Cysts are lined by low cuboidal epithelium surrounded by spindle cells and may be filled with proteinaceous or sanguineous fluid.1 Primitive and dysplastic components as well as immature cartilage or immature glomeruli can be seen. MCDK is classified as infundibulopelvic type with atresia of renal pelvis and ureter versus the less common hydronephrotic type that only involves atresia of upper ureter.46 The contralateral kidney may show compensatory hypertrophy.
Evaluation and Diagnosis
Currently, MCDK is discovered on prenatal US most commonly, whereas evaluation of a palpable mass in a neonate is the next most common presentation.47
A diagnosis of MCDK must be differentiated from hydronephrosis due to other conditions like UPJ obstruction. Notably, in MCDK, cysts are noncommunicating and larger cysts appear laterally. In hydronephrosis, renal parenchyma surrounds the central cystic structure and the typical reniform shape is maintained.48 Figure 3 and Figure 4 help demonstrate the challenges of differentiating MCDK from hydronephrosis on ultrasound. Figure 5 illustrates noncommunicating cysts of various sizes, typical of MCDK. To further clarify, a radionuclide scan can be performed which would show uptake with hydronephrosis and typically minimal or no uptake in the setting of MCDK.49
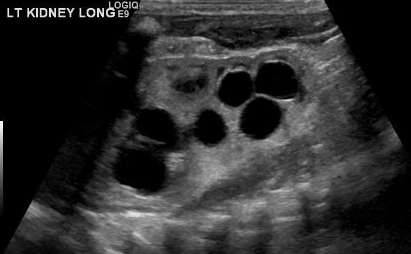
Figure 3 Renal ultrasound demonstrating what could be several renal cysts in an MCDK versus pelvicaliectasis/hydronephrosis (HDN).
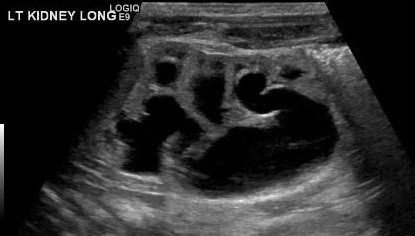
Figure 4 Another renal ultrasound image for this same patient, demonstrating continuity between pelvis and calyces, consistent with pelvicaliectasis/HDN.
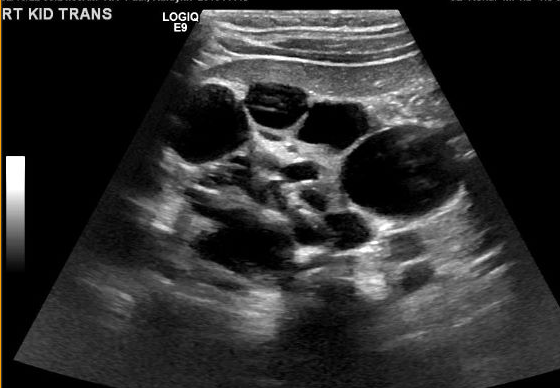
Figure 5 Renal ultrasound demonstrating multiple noncommunicating cysts of different sizes, consistent with MCDK.
Treatment Options, Their Outcomes, and Complications
The natural history of MCDK involves involution over time with possible complications including hypertension and malignancy.5 However, the risk of the latter may be low.50 Historically, affected kidneys were surgically removed out of concern for rare cases of malignant degeneration, but there has been a shift toward non-operative care with monitoring ultrasonography of both kidneys. Indications for surgery include changes suspicious for development of Wilms’ tumor, mass effect, pain, hypertension, and parental preference.5 Patients with MCDK and a normal contralateral kidney should be reassured of anticipated long-term normal renal function, barring injury to their contralateral kidney. They should also be counseled on the importance of preventive maintenance measures, like using protective gear when engaging in sports, and healthy lifestyle choices (diet and exercise) to mitigate other causes of renal disease (like hypertension and diabetes). To characterize risk of chronic kidney disease development, surveillance can additionally include contralateral kidney imagining, voiding cystourethrogram to screen for VUR, blood pressure monitoring, and urinalysis for proteinuria.51
Medullary Sponge Kidney
Medullary sponge kidney (MSK) is characterized by distal collecting duct dilation, cysts, and diverticula all restricted to the medullary pyramids.1
Epidemiology
MSK is primarily found in young adulthood, but may also present in children.52 As many individuals with MSK are asymptomatic, the incidence is not clearly defined. It is more common in people with a calcium stone history and has been associated with congenital syndromes, such as Beckwith-Wiedemann syndrome, Ehlers-Danlos syndrome, anodontia, and Caroli disease.1
Pathogenesis
MSK is largely considered nonhereditary, however, there have been cases that seem to be inherited in an autosomal dominant fashion supporting a hypothesis that MSK disrupts the ureteric bud and metanephric blastema interface.52
Dilated intrapapillary collecting duct and medullary cysts contain calcified or desquamated material, give the kidney a sponge-like appearance, and are bilateral in 70% of cases.1 Cysts are contiguous with collecting tubules and lined by collecting duct epithelium.53
Evaluation and Diagnosis
Diagnosis may be based on urographic features of enlarged kidneys with occasional calcification especially at the papillae, enlarged papillary tubules that fill with contrast, and papillary contrast blush with residual medullary opacification.54 Liver evaluation can clarify between MSK and ADPKD, if diagnosis is unclear.1
Treatment Options, Their Outcomes, and Complications
Management is primarily dictated by stone or infection treatment. Stone prevention is followed, including increased fluid intake, low sodium diet, thiazides, and potassium citrate.52 Complications associated with MSK are related to the increased incidence of nephrolithiasis and its associated morbidity.
Nephronophthisis
Nephronophthisis (NPHP) is a recessively inherited common cause of chronic kidney disease in children, often paired with medullary cystic kidney disease (MCKD) as they have similar radiographic and histologic appearance; however MCKD is inherited in an autosomal dominant fashion, has delayed presentation (adulthood), and lacks extrarenal involvement.5
Epidemiology
NPHP is classified as infantile (1 year of age), juvenile (13 years of age), or adolescent (19 years of age) based on the genetic defect present. Infantile NPHP leads to ESRD by 1–3 years of age and is associated with NPHP2 mutations (also referred to as NPHP type 2). Juvenile NPHP, the most common manifestation, leads to ESRD at 10–13 years and is most often associated with NPHP1 mutation (also referred to as NPHP type 1). Lastly, adolescent NPHP is associated with NPHP3 mutations (also referred to as NPHP type 3).5,55
Pathogenesis
Although it can occur sporadically, NPHP is typically inherited in an autosomal recessive fashion with homozygous deleterious mutations of NPHP1 on chromosome 2 as the most common cause.56 There are many genes known to be associated with NPHP (see (Table 2)5
Table 2 Genetic heterogeneity of NPHP.5
| Type | Chromosome location | Gene mutated |
|---|---|---|
| NPH type 1 | 2q13 | NPHP1 |
| NPH type 2 | 9q22 | NPHP2 |
| NPH type 3 | 3q22 | NPHP3 |
| NPH type 4 | 1p36 | NPHP4 |
| NPH type 5 | 3q21 | NPHP5 |
| PH type 6 | 12q21 | NPHP6 |
| NPH type 7 | 16p | NPHP7 |
| NPH type 8 | 16q | NPHP8 |
| NPH type 9 | 17q11 | NPHP9 |
| NPH type 10 | 1q44 | SDCCAG8 |
| PH type 11 | 8q22 | MKS3 |
| AHI1 | 6q23 | AHI1 |
Gross evaluation of NPHP kidneys may vary based on type but tend to be small in size with a granular surface. Histologically, they display interstitial fibrosis and mononuclear cell infiltration, variable tubule changes including atrophy, thickening, and lamination of basement membranes.57 Cysts in NPHP arise from distal convoluted tubule and collecting ducts.57
Evaluation, Diagnosis, and Complications
Problems with urine concentrating ability may lead to polyuria and polydipsia (dehydration, nocturia) with resultant renal sodium wasting; symptoms of CKD such as fatigue, anorexia, growth retardation; as well as symptoms of renal osteodystrophy. Anemia may be present out of proportion to renal failure. HTN is less common except in type 2 NPHP.5,58 Extrarenal involvement includes hepatic and portal fibrosis with hepatomegaly as well as many others: Senior-Løken syndrome (NPHP and tapetoretinal degeneration), skeletal abnormalities, mental retardation, cerebellar ataxia, situs inversus, cardiac malformation, or other less common syndromes.5
Ultrasonography is a common method for identification, but may not detect all cases; thin-section computed tomography (CT) may capture such cases.58 Genetic testing is available for diagnosis as well as renal histology when imaging is inconclusive. Liver and ocular screening is recommended for hepatic fibrosis and Senior-Løken syndrome.5
Treatment Options and Their Outcomes
There are few effective treatments available for NPHP. Current management includes supportive and preventive management for CKD. Transplantation in setting of kidney failure is preferred, as tubular injury does not occur in the donor kidney.13
Multilocular Cysts
Multilocular cysts (cystic nephroma) is a benign neoplastic growth occurring in adults and children that exists on a spectrum with Wilms’ tumor.
Epidemiology
The majority of cases occur before 4 years of age or after 30 years of age, with a male predominance in the pediatric population.59
Pathogenesis
Lesions are bulky, non-communicating, well encapsulated by fibrous tissue, may contain embryonic tissue, and may compress normal renal parenchyma.5
Evaluation, Diagnosis, and Treatment Options
Multilocular cysts in children most commonly present with a palpable abdominal mass.60 Evaluation is performed with ultrasound or CT scan, however, due to the difficulty in sometimes discerning between multilocular cysts and cystic Wilms’ tumor based purely upon imaging, ultimate diagnosis may be made via histopathologic examination following nephrectomy or partial nephrectomy.60
Acquired Cystic Kidney Diseases
Acquired cystic kidney disease (ACKD) describes the bilateral cystic changes of the kidney in the setting of ESRD and azotemia and not inherited renal cystic disease.1
Epidemiology
Cysts are found in 10% of patients with ESRD, increasing to 44% and 60% at 3 and 5 years after initiation of dialysis.1
Pathogenesis
Etiology is thought to be toxin mediated, accumulation of growth factors, or tubular obstruction due to fibrosis, oxalate crystals, vascular occlusion or ischemia.1 Kidneys are bilaterally affected and usually smaller in size than normal, while histology shows flat to cuboidal epithelium similar to distal tubular epithelium.4
Evaluation, Diagnosis, Treatment, and Complications
Most patients with cysts are asymptomatic but may present with pain and/or hematuria due to spontaneous bleeding into cysts. Ultrasonography is commonly used for diagnosis and monitoring while CT or MRI may more readily identify cysts, however in ESRD patients imaging with IV contrast should be carefully managed due to the nephrotoxic effects of contrast agents.1 The main complications are hemorrhage and neoplastic transformation.4 Surveillance ultrasonography is recommended with nephrectomy in a setting suspicious for neoplasia.5 Thus treatment is managed conservatively with symptom management and modulation of screening schedules or dialysis regimens.
Syndromes Associated with Cystic Kidney Disease
Renal cysts can be seen in various syndromes, the most common of which may include autosomal dominant tuberous sclerosis and Von Hippel-Lindau. Others of autosomal recessive inheritance patterns include Meckel syndrome, Jeune asphyxiating thoracic dystrophy, and Zellweger cerebrohepatorenal syndrome.1
Tuberous Sclerosis
Tuberous sclerosis is diagnosed by presence of either two major criteria or one major plus two minor criteria (see Table 3).61 Multiple renal cysts is included as a minor criteria.61,62 Renal involvement is a significant cause of morbidity and mortality in tuberous sclerosis.63
Table 3 Major and minor criteria for tuberous sclerosis complex.61
| Major Criteria | Minor Criteria |
|---|---|
| Hypomelanotic macules (≥3, at least 5-mm diameter) | “Confetti” skin lesions |
| Angiofibromas (≥3) or fibrous cephalic plaque | Dental enamel pits (>3) |
| Ungual fibromas (≥2) | Intraoral fibromas (≥2) |
| Shagreen patch | Retinal achromic patch |
| Multiple retinal hamartomas | Multiple renal cysts |
| Cortical dysplasias | Nonrenal hamartomas |
| Subependymal nodules | |
| Subependymal giant cell astrocytoma | |
| Cardiac rhabdomyoma | |
| Lymphangioleiomyomatosis (LAM) | |
| Angiomyolipomas (≥2) |
Von Hippel-Lindau Syndrome
Von Hippel-Lindau syndrome similarly has many manifestations, of which renal cell carcinoma and renal cysts are common, but infrequently seen in children.64
Meckel syndrome
This may present with a variety of manifestations including cysts. Cystic dysplasia is always present.5,65
Conclusions
Fluid-filled renal cysts are found in a large, heterogenous group of renal cystic diseases. Although they share a common manifestation as renal cysts, this group of diseases are differentiated by many factors: pattern of inheritance, age of onset, associated renal or extrarenal abnormalities, clinical course, histological differences, appearance on imaging, among others. Accurate identification of a disease process may be challenging with overlapping symptomatology. Imagining techniques, especially ultrasonography, has long been a primary mode of identifying many of these diseases. Genetic testing options are becoming more common and accessible.
Continued research leads to basic science and clinical advancements, helping to identify new diseases, pathophysiology, diagnostic modalities, and treatment options.
Key Points
- Distinction between a simple cyst and a calyceal diverticulum may be difficult, however of particular importance as a calyceal diverticulum is much more likely to cause symptoms and require intervention. Diagnosis can be facilitated using cross-sectional imaging with IV contrast and delayed phase images.
- It is also of importance to distinguish between MCDK and severe hydronephrosis, as in the setting of ureteropelvic junction (UPJ) obstruction. The management of MCDK is largely observational whereas there is a higher likelihood that intervention may be necessary for hydronephrosis or UPJ obstruction. Diagnosis can be facilitated using radionuclide imaging.
- Management of certain cystic kidney diseases, such as ADPKD and ARPKD, should be done in partnership with nephrology to facilitate optimal medical-renal care.
- Characterization of cysts by prenatal imaging often help differentiate between possible diseases (see Figure 6 below).13

Figure 6 Differentiation of cysts by prenatal imaging.13
Suggested Readings
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
References
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- Glassberg KI, Stephens FD, Lebowitz RL. Renal dysgenesis and cystic disease of the kidney: a report of the Committee on Terminology, Nomenclature and Classification, Section on Urology, American Academy of Pediatrics. J Urol Oct 1987; 138 (4 Pt 2): 1085–1092. DOI: 10.1016/s0022-5347(17)43510-5.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364 (16): 1533–1543. DOI: 10.1056/NEJMra1010172.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Cole BR, Conley SB, Stapleton FB. Polycystic kidney disease in the first year of life. J Pediatr Nov 1987; 111 (5): 693–699. DOI: 10.1016/s0022-3476(87)80244-5.
- Gabow PA. Polycystic kidney disease: clues to pathogenesis. Kidney Int Dec 1991; 40 (6): 989–996. DOI: 10.1038/ki.1991.306.
- Proesmans W, Damme B, Casaer P, Marchal G. Autosomal dominant polycystic kidney disease in the neonatal period: association with a cerebral arteriovenous malformation. Pediatrics Dec 1982; 70 (6): 971–975. DOI: 10.1542/peds.70.6.971.
- Ong AC, Wheatley DN. Polycystic kidney disease–the ciliary connection. Lancet Mar 2003; 361 (9359): 774–776. DOI: 10.1016/S0140-6736(03)12662-1.
- Rossetti S, Consugar MB, Chapman AB. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol Jul 2007; 18 (7): 2143–2160. DOI: 10.1681/ASN.2006121387.
- Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol Oct 2004; 15 (10): 2528–2536. DOI: 10.1097/01.ASN.0000141055.57643.E0.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet Apr 1994; 343 (8901): 824–827. DOI: 10.1016/s0140-6736(94)92026-5.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Grantham JJ. Polycystic kidney disease: hereditary and acquired. Adv Intern Med 1993; 38: 409–420.
- Schrier R, McFann K, Johnson A. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol Jul 2002; 13 (7): 1733–1739. DOI: 10.1097/01.asn.0000018407.60002.b9.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med Oct 1990; 323 (16): 1091–1096. DOI: 10.1056/NEJM199010183231602.
- Alzarka B, Morizono H, Bollman JW, Kim D, Guay-Woodford LM. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Front Pediatr 2017; 5 (80). DOI: 10.3389/fped.2017.00080.
- Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers 2018; 4 (1). DOI: 10.1038/s41572-018-0047-y.
- Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatr Nephrol Jan 1989; 3 (1): 43–49. DOI: 10.1007/BF00859625.
- Ward CJ, Hogan MC, Rossetti S. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet Mar 2002; 30 (3): 259–269. DOI: 10.1038/ng833.
- Onuchic LF, Furu L, Nagasawa Y. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet May 2002; 70 (5): 1305–1317. DOI: 10.1086/340448.
- Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol Jun 1997; 11 (3): 302–306. DOI: 10.1007/s004670050281.
- Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics Sep 2014; 134 (3). DOI: 10.1542/peds.2013-3646.
- Blazer S, Zimmer EZ, Blumenfeld Z, Zelikovic I, Bronshtein M. Natural history of fetal simple renal cysts detected in early pregnancy. J Urol 1999; 162 (3 Pt 1): 812–814. DOI: 10.1097/00005392-199909010-00066.
- McHugh K, Stringer DA, Hebert D, Babiak CA. Simple renal cysts in children: diagnosis and follow-up with US. Radiology Feb 1991; 178 (2): 383–385. DOI: 10.1148/radiology.178.2.1987597.
- Bosniak MA. The current radiological approach to renal cysts. Radiology Jan 1986; 158 (1): 1–10. DOI: 10.1148/radiology.158.1.3510019.
- Lee J, Darcy M. Renal cysts and urinomas. Semin Intervent Radiol. Dec 2011; 28 (4): 380–391. DOI: 10.1055/s-0031-1296080.
- Steinhardt GF, Slovis TL, Perlmutter AD. Simple renal cysts in infants. Radiology May 1985; 155 (2): 349–350. DOI: 10.1148/radiology.155.2.3885305.
- Gabow PA, Kimberling WJ, Strain JD, Manco-Johnson ML, Johnson AM. Utility of ultrasonography in the diagnosis of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol Jan 1997; 8 (1): 105–110. DOI: 10.1681/ASN.V81105.
- Peard L, Gargollo P, Grant C. Validation of the modified Bosniak classification system to risk stratify pediatric cystic renal masses: An international, multi-site study from the pediatric urologic oncology working group of the societies for pediatric urology. J Pediatr Urol 2022; 18 (2). DOI: 10.1016/j.jpurol.2021.12.001.
- Bayram MT, Alaygut D, Soylu A, Serdaroğlu E, Cakmakçı H, Kavukçu S. Clinical and radiological course of simple renal cysts in children. Urology Feb 2014; 83 (2): 433–437. DOI: 10.1016/j.urology.2013.08.055.
- Karmazyn B, Tawadros A, Delaney LR. Ultrasound classification of solitary renal cysts in children. J Pediatr Urol Jun 2015; 11 (3). DOI: 10.1016/j.jpurol.2015.03.001.
- O’Kelly F, McAlpine K, Abdeen N, Keays MA, Leonard MP, Guerra LA. The Prevalence, Clinicodemographics, and Outcomes of Incidental and Symptomatic Renal Cysts in a Pediatric Cohort Undergoing Ultrasonography. J Urol 2019; 202 (2): 394–399. DOI: 10.1097/JU.0000000000000264.
- Rediger C, Guerra LA, Keays MA. Renal cyst evolution in childhood: a contemporary observational study. J Pediatr Urol Apr 2019; 15 (2). DOI: 10.1016/j.jpurol.2019.01.006.
- Wulfsohn MA. Pyelocaliceal diverticula. J Urol Jan 1980; 123 (1): 1–8. DOI: 10.1016/s0022-5347(17)55748-1.
- Estrada CR, Datta S, Schneck FX, Bauer SB, Peters CA, Retik AB. Caliceal diverticula in children: natural history and management. J Urol Mar 2009; 181 (3): 1311. DOI: 10.1016/j.juro.2008.10.043.
- Waingankar N, Hayek S, Smith AD, Okeke Z. Calyceal diverticula: a comprehensive review. Rev Urol 2014; 16 (1): 29–43.
- Sahin H, Sarioglu FC, Alaygut D, Akdogan AI, Pekcevik Y. Differentiation of simple renal parenchymal cyst and calyceal diverticulum. Pediatr Int May 2020; 62 (5): 615–623. DOI: 10.1111/ped.14127.
- Casale P, Grady RW, Feng WC, Joyner BD, Mitchell ME. The pediatric caliceal diverticulum: diagnosis and laparoscopic management. J Endourol Sep 2004; 18 (7): 668–671. DOI: 10.1089/end.2004.18.668.
- Long CJ, Weiss DA, Kolon TF, Srinivasan AK, Shukla AR. Pediatric calyceal diverticulum treatment: An experience with endoscopic and laparoscopic approaches. J Pediatr Urol Aug 2015; 11 (4). DOI: 10.1016/j.jpurol.2015.04.013.
- Sripathi V, Mitra A, Padankatti RL, Ganesan T. Robotic treatment of a type 2 calyceal diverticulum in a child: is suture closure and marsupialisation enough for a good outcome? J Robot Surg Dec 2018; 12 (4): 727–730. DOI: 10.1007/s11701-017-0758-1.
- Ding X, Xu ST, Huang YH. Management of symptomatic caliceal diverticular calculi: Minimally invasive percutaneous nephrolithotomy versus flexible ureterorenoscopy. Chronic Dis Transl Med Dec 2016; 2 (4): 250–256. DOI: 10.1016/j.cdtm.2016.11.016.
- Monga M, Smith R, Ferral H, Thomas R. Percutaneous ablation of caliceal diverticulum: long-term followup. J Urol Jan 2000; 163 (1): 28–32. DOI: 10.1016/s0022-5347(05)67965-7.
- Gilbert-Barness E, Potter EL. Respiratory system. 2nd ed., DOI: 10.1001/jama.1997.03540320078045.
- D’Alton M, Romero R, Grannum P, DePalma L, Jeanty P, Hobbins JC. Antenatal diagnosis of renal anomalies with ultrasound. IV Bilateral Multicystic Kidney Disease Am J Obstet Gynecol Mar 1986; 154 (3): 532–537. DOI: 10.1016/0002-9378(86)90597-1.
- Kalyoussef E, Hwang J, Prasad V, Barone J. Segmental multicystic dysplastic kidney in children. Urology Nov 2006; 68 (5). DOI: 10.1016/j.urology.2006.06.024.
- Felson B, Cussen LJ. The hydronephrotic type of unilateral congenital multicystic disease of the kidney. Semin Roentgenol Apr 1975; 10 (2): 113–123. DOI: 10.1016/0037-198x(75)90035-8.
- Welch TR, Wacksman J. The changing approach to multicystic dysplastic kidney in children. J Pediatr Jun 2005; 146 (6): 723–725. DOI: 10.1016/j.jpeds.2005.02.027.
- Sanders RC, Hartman DS. The sonographic distinction between neonatal multicystic kidney and hydronephrosis. Radiology Jun 1984; 151 (3): 621–625. DOI: 10.1148/radiology.151.3.6718720.
- Roach PJ, Paltiel HJ, Perez-Atayde A, Tello RJ, Davis RT, Treves ST. Renal dysplasia in infants: appearance on 99mTc DMSA scintigraphy. Pediatr Radiol 1995; 25 (6): 472–475. DOI: 10.1007/BF02019071.
- Narchi H. Risk of Wilms’ tumour with multicystic kidney disease: a systematic review. Arch Dis Child Feb 2005; 90 (2): 147–149. DOI: 10.1136/adc.2004.051243.
- Mansoor O, Chandar J, Rodriguez MM. Long-term risk of chronic kidney disease in unilateral multicystic dysplastic kidney. Pediatr Nephrol Apr 2011; 26 (4): 597–603. DOI: 10.1007/s00467-010-1746-0.
- Gambaro G, Danza FM, Fabris A. Medullary sponge kidney. Curr Opin Nephrol Hypertens. Jul 2013; 22 (4): 421–426. DOI: 10.1097/MNH.0b013e3283622b86.
- Bernstein J. The classification of renal cysts. Nephron 1973; 11 (2): 91–100. DOI: 10.1159/000180222.
- Gedroyc WM, Saxton HM. More medullary sponge variants. Clin Radiol Jul 1988; 39 (4): 423–425. DOI: 10.1016/s0009-9260(88)80292-7.
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol Jan 2009; 20 (1): 23–35. DOI: 10.1681/ASN.2008050456.
- Hildebrandt F, Otto E, Rensing C. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet Oct 1997; 17 (2): 149–153. DOI: 10.1038/ng1097-149.
- Sherman FE, Studnicki FM, Fetterman G. Renal lesions of familial juvenile nephronophthisis examined by microdissection. Am J Clin Pathol Apr 1971; 55 (4): 391–400. DOI: 10.1093/ajcp/55.4.391.
- Elzouki AY, al-Suhaibani H, Mirza K, al-Sowailem AM. Thin-section computed tomography scans detect medullary cysts in patients believed to have juvenile nephronophthisis. Am J Kidney Dis Feb 1996; 27 (2): 216–219. DOI: 10.1016/s0272-6386(96)90543-0.
- Eble JN, Bonsib SM. Extensively cystic renal neoplasms: cystic nephroma, cystic partially differentiated nephroblastoma, multilocular cystic renal cell carcinoma, and cystic hamartoma of renal pelvis. Semin Diagn Pathol Feb 1998; 15 (1): 2–20.
- Castillo OA, Boyle ET, Kramer SA. Multilocular cysts of kidney. A study of 29 patients and review of literature. Urology Feb 1991; 37 (2): 156–162. DOI: 10.1016/0090-4295(91)80214-r.
- Northrup H, Krueger DA, ITSCC G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol Oct 2013; 49 (4): 243–254. DOI: 10.1016/j.pediatrneurol.2013.08.001.
- Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol Dec 1998; 13 (12): 624–628. DOI: 10.1177/088307389801301206.
- Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc Aug 1991; 66 (8): 792–796. DOI: 10.1016/s0025-6196(12)61196-3.
- Maddock IR, Moran A, Maher ER. A genetic register for von Hippel-Lindau disease. J Med Genet Feb 1996; 33 (2): 120–127. DOI: 10.1136/jmg.33.2.120.
- Salonen R. The Meckel syndrome: clinicopathological findings in 67 patients. Am J Med Genet Aug 1984; 18 (4): 671–689. DOI: 10.1002/ajmg.1320180414.
Last updated: 2025-09-25 12:10