
40: DSD—当前认识、诊断评估与治疗
阅读本章大约需要 8 分钟。
简介与术语
性发育障碍/差异(DSD,也称为间性)是先天性状况,其中染色体性别、性腺性别或表型性别与通常所见的典型男性或女性不同。2006年通过 “间性障碍管理共识声明” 引入了新的、范围更广的DSD分类。1 这种较新的分类涵盖了广泛的疾病,包括先天性肾上腺皮质增生(CAH)、卵睾性DSD、雄激素不敏感综合征(AIS)。2006年的共识定义比此前所称的间性状态更加宽泛,还包括解剖学异常,如泄殖腔外翻、膀胱外翻和阴道缺如,以及不导致非典型外生殖器的染色体异常(如克氏综合征 [47, XXY])。表 1 概述了此前的命名法以及2006年共识声明提出的术语更新建议。鉴于DSD ‘伞式’范畴下所涵盖的疾病范围广泛,个体化、多学科的照护方式至关重要。
表 1 修订的 DSD 命名法。来源:Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
| 既往术语 | 建议术语 |
|---|---|
| 两性畸形 | DSD |
| 男性假两性畸形 | 46,XY DSD |
| 女性假两性畸形 | 46,XX DSD |
| 真两性畸形 | 卵睾性 DSD |
| 混合性性腺发育不良 | 混合性性腺发育不良(未更改) |
| XX 男性或 XX 性逆转 | 46,XX 睾丸性 DSD |
| XY 性逆转 | 46,XY 完全性腺发育不良 |
自从新的 DSD 术语被引入以来,它几乎已被医学界普遍采纳。在受 DSD 情况影响的个体中,术语偏好存在差异,对于究竟哪些情况应被视为 DSD 仍缺乏共识。2,3,4 尤其是,先天性肾上腺皮质增生症(CAH)社群中的一些成员将自身归类为患有内分泌疾病,而非 DSD/间性情况。5 对于哪些近端尿道下裂个体应被视为“DSD”,也尚不明确。6 为 DSD 情况提供照护的临床医生应了解该命名法的演变及其争议,并在个体医疗接触中使用患者所偏好的术语。出于本章的目的,将采用当前医学界认可的术语。
胚胎学
内/外生殖器的典型发育
所有发育中的胎儿在起始阶段相同。在妊娠早期数周内,就已存在用于男性或女性发育及其变异的解剖学基础(表 2),包括性腺嵴、沃尔夫管(中肾管)和苗勒管(副中肾管)、泄殖腔及其后续形成的泌尿生殖窦、生殖结节和阴唇阴囊隆起。图 1显示了负责将未分化性腺发育为睾丸或卵巢的基因。
表 2 胚胎学前体及典型的男性/女性结构.
| 胚胎学结构 | 女性典型结构 | 男性典型结构 |
|---|---|---|
| 性腺嵴 | 卵巢 | 睾丸 |
| 沃尔夫管(中肾管) | 卵巢旁体、卵巢上体、加特纳管囊肿 | 输精管、副睾、精囊 |
| 苗勒管(旁中肾管) | 输卵管、子宫、阴道近端 | 睾丸阑尾、前列腺小囊 |
| 泄殖腔及其后形成的泌尿生殖窦 | 膀胱、阴道远端、尿道 | 膀胱、前列腺、尿道 |
| 生殖结节 | 阴蒂 | 阴茎 |
| 阴唇阴囊隆起 | 阴唇复合体 | 阴囊 |
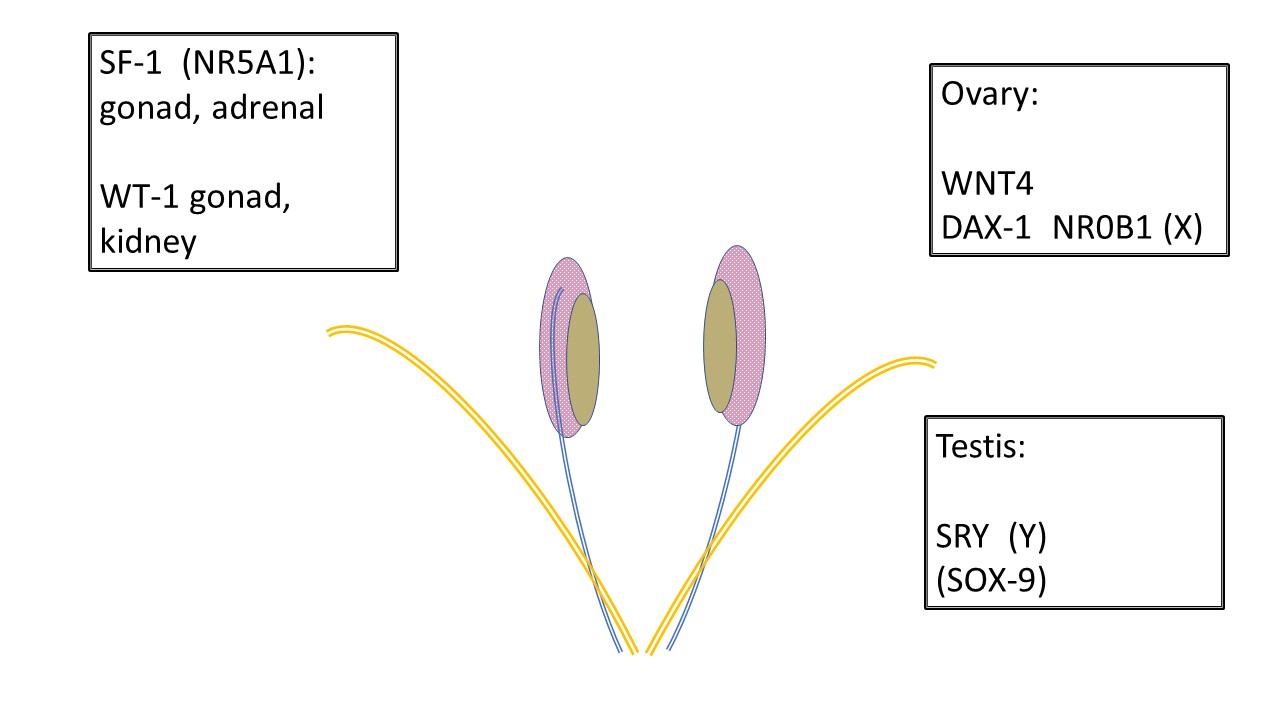
图 1 未分化性腺的发育。 所有性腺起初相同。原始生殖细胞在 6 周前迁移至性腺嵴,支持性腺发育的基础结构还进一步受多种基因影响。SF-1 和 WT-1 影响性腺发育,以及随后向沃尔夫管(中肾管-蓝色)和米勒管(副中肾管-橙色)的内分泌信号传递。注意中肾(粉色)、中肾管与未分化性腺(棕褐色)之间的关系。WNT4 是卵巢决定基因。DAX-1 被认为是 “抗睾丸基因”。DAX-1 的重复会导致 XY 性逆转。位于 Y 染色体上的 SRY 是睾丸决定基因。SOX-9 支持塞尔托利细胞发育,并且由于其同源性,在缺乏 SRY 时仍可参与睾丸发育。
典型的男性发育(图2)在孕期非常早期即由Y染色体,具体为SRY,启动并决定睾丸的发育。WT-1、SF-1参与睾丸和卵巢的发育,而SOX-9参与Sertoli细胞的分化。随后在孕约8周时,睾丸的莱迪希细胞(Leydig细胞)开始产生睾酮,Sertoli细胞产生抗苗勒管激素(AMH),抑制苗勒管成熟为子宫、输卵管和阴道上段。睾酮决定生殖结节的早期生长,并促使沃尔夫管发育为输精管、附睾和精囊。沃尔夫管和苗勒管系统的发育或缺如是通过旁分泌而非全身性作用发生的,这解释了当性腺组成不对称时导管结构的不对称发育。睾酮在多种组织中经2型5α-还原酶转化为更强效的二氢睾酮(DHT),该过程负责阴茎的进一步成熟、泌尿生殖窦和尿道板的成管化,并在妊娠约14周时完成阴囊的闭合。随后,随着胎儿雄激素逐渐减弱以及体躯生长的推进,阴茎结构继续增长。
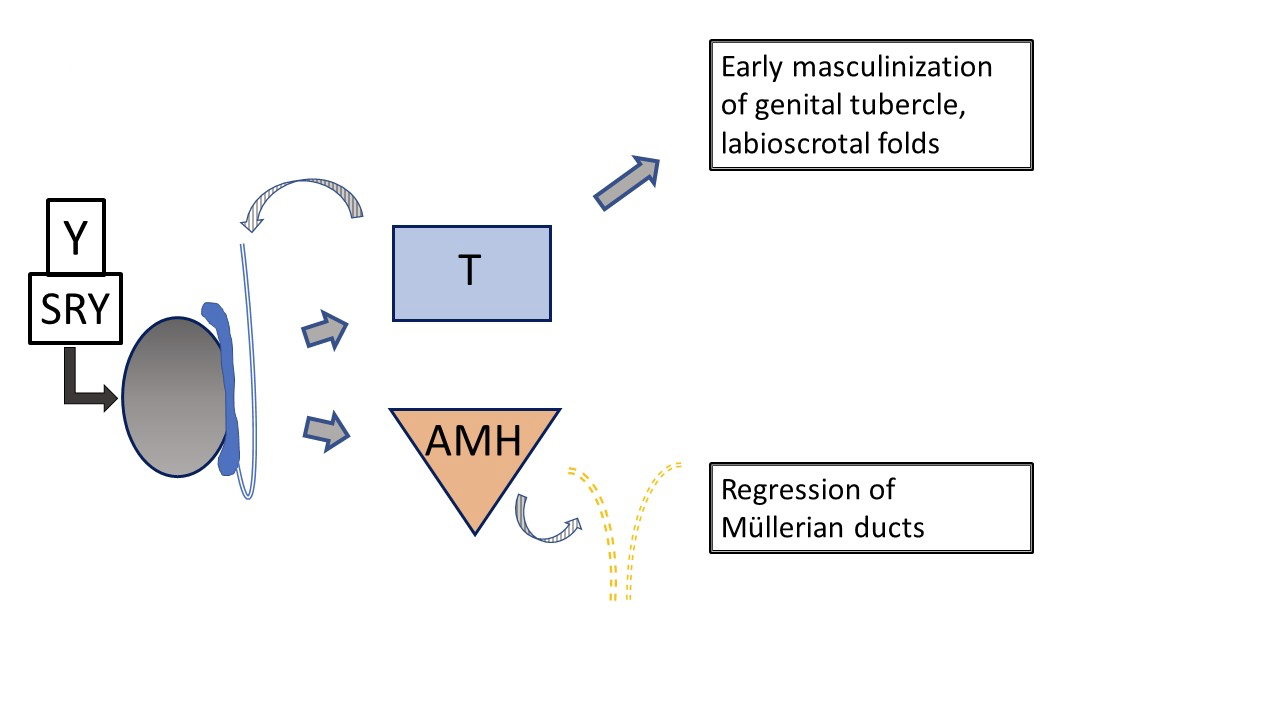
图 2 胎儿睾丸影响下男性外生殖器和内生殖道的典型早期发育. 胎儿睾丸产生的睾酮和 AMH 导致生殖结节的早期男性化以及苗勒管的退化. 外生殖器和尿道的后续发育受睾酮转化为二氢睾酮的影响. (此处未示出, 见图 4).
典型的女性发育(图3)通常被认为是在默认情况下发生,因为它不受睾丸产生的激素的主导。卵巢的确定并未在生殖器的胚胎学发育中占据突出位置。低水平的雄激素和AMH使生殖结节发育为阴蒂、阴唇阴囊褶发育为阴唇复合体、远端尿生殖窦发育为尿道和阴道。成对的苗勒管迁移并融合形成子宫。它们共同刺激尿生殖窦上的阴窦球,启动下段阴道的发育。
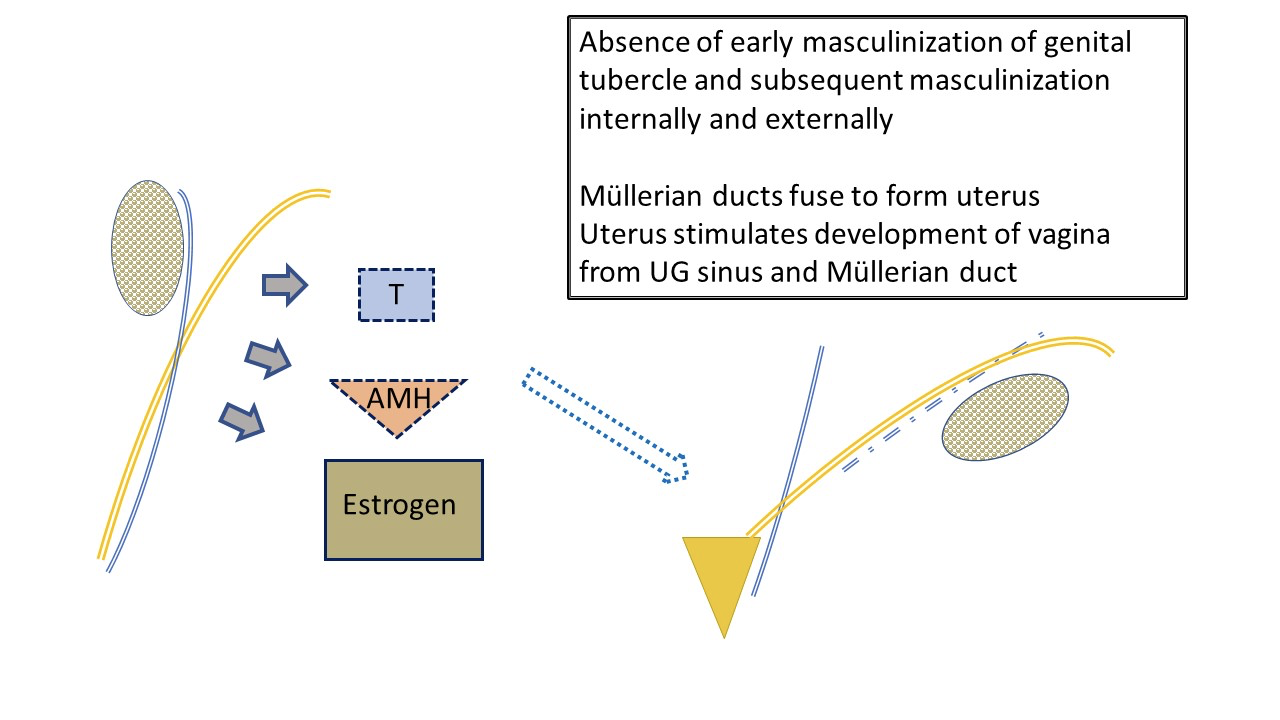
图 3 受胎儿卵巢影响的女性外生殖器和内生殖道的典型早期发育。卵巢分泌的抗缪勒氏管激素(AMH)和雄激素水平较低,导致典型女性解剖结构的发育:子宫和输卵管、阴道上段。与雄激素和AMH处于极低水平的环境相比,胎儿雌激素的产生对女性谱系发育的影响较小。阴唇会受到母体雌激素的影响。另请注意,输尿管(由中肾管分支而来)与输卵管之间最终呈现 “water under the bridge” 的关系。注意与输卵管相关的中肾管残余。
可能导致 DSD 的不同通路
发育差异可能通过非典型的方式发生:
- 性腺的遗传决定 (例如,混合性性腺发育不全、卵睾性 DSD)
- 激素产生 (例如,5α-还原酶缺乏症、3β-羟类固醇脱氢酶缺乏症、21-羟化酶缺乏 CAH,或黄体生成素 [LH] 受体突变)
- 激素作用 (例如,完全或部分性 AIS)
- 前体组织发育/泄殖腔分隔的变异 (例如,Mayer-Rokitansky-Küster-Hauser [MRKH] 综合征、泄殖腔外翻、泄殖腔畸形)
即使具有46,XX或46,XY核型,发育也并不能确保沿典型的女性或男性方向进行。仅就46,XY而言,由于改变睾丸决定或功能的Y染色体突变、导致发育中睾丸发育不良的体细胞突变、睾酮生物合成或转化的差异,以及影响雄激素受体功能的X连锁突变,亦可出现诸多差异。外部表型、内部发育、自发进入青春期或生育力的潜能,以及在性别谱系中的认同均可能受到影响。在考虑下文所述各类DSD状况的特征时,需要注意的是,激素的生成与作用呈谱系变化。因此,受体功能缺陷或激素及转化酶生成的不足并不一定是完全性缺陷。即便在具有相同诊断的个体之间,表型也可能差异很大。
病理生理学
内外生殖器发育的变异受性染色体和体细胞突变、由此导致的性腺功能以及组织反应所主导。当由于已知或尚未鉴定的遗传突变导致受体功能或相关酶的存在情况异常时,对靶器官的影响可以是完全的(无明显功能)或部分的。表型因此而异。例如,雄激素不敏感的各种表型(完全性 CAIS、部分性 PAIS 和轻度 MAIS:生殖器表型谱从典型女性—不明确—典型男性)可由位于 X染色体上的雄激素受体基因的多种不同突变所致(X连锁隐性遗传)。表 3 展示了一个按核型划分的 DSD 分类系统。
表 3 DSD 分类示例
| 性染色体 DSD | 46,XY DSD | 46,XX DSD |
|---|---|---|
| 45,X(特纳综合征及其变异) | 性腺(睾丸)发育异常: (1) 完全性腺发育不全(Swyer 综合征); (2) 部分性腺发育不全; (3) 性腺退化;以及 (4) 卵睾型 DSD |
性腺(卵巢)发育异常: (1) 卵睾型 DSD; (2) 睾丸型 DSD(例如,SRY+,SOX9 重复);以及 (3) 性腺发育不全 |
| 47,XXY(克氏综合征及其变异) | 雄激素合成或作用异常: (1) 雄激素生物合成缺陷(例如,17-羟类固醇脱氢酶缺乏、5RD2 缺乏、StAR 突变); (2) 雄激素作用缺陷(例如,CAIS、PAIS); (3) 黄体生成素受体缺陷(例如,Leydig 细胞发育不全、发育缺如); 以及 (4)抗米勒管激素及抗米勒管激素受体的异常(持续性米勒管综合征) |
雄激素过多: (1) 胎儿期(例如,21-羟化酶缺乏、11-羟化酶缺乏); (2) 胎儿-胎盘来源(芳香化酶缺乏、POR [P450 氧化还原酶]);以及 (3) 母体(黄体瘤、外源性等) |
| 45,X/46,XY(MGD,卵睾型 DSD) | 其他(例如,泄殖腔外翻、阴道闭锁、MURCS [米勒管、肾、颈胸部体节异常]、其他综合征) | |
| 46,XX/46,XY(嵌合体,卵睾型 DSD) |
46,XX 性发育差异
最常见的46,XX DSD形式是CAH,其中肾上腺酶学功能受损。图 4 显示了典型的肾上腺类固醇合成级联,始于胆固醇。终产物为醛固酮、皮质醇和性类固醇。该级联的性类固醇支路以雄烯二酮为终点,后者可转化为少量睾酮。睾丸的 Leydig 细胞中睾酮的合成同样始于胆固醇。在CAH中,酶功能降低导致矿物皮质激素(醛固酮)和糖皮质激素(皮质醇)减少,并分流至性类固醇的合成。其结果是具有46,XX核型的胎儿发生男性化。尽管最容易记住的是酶活性完全缺失以及下游肾上腺类固醇产物缺如的情形,但实际上功能可能仅部分受损,并因而相应影响下丘脑-垂体轴的反馈。
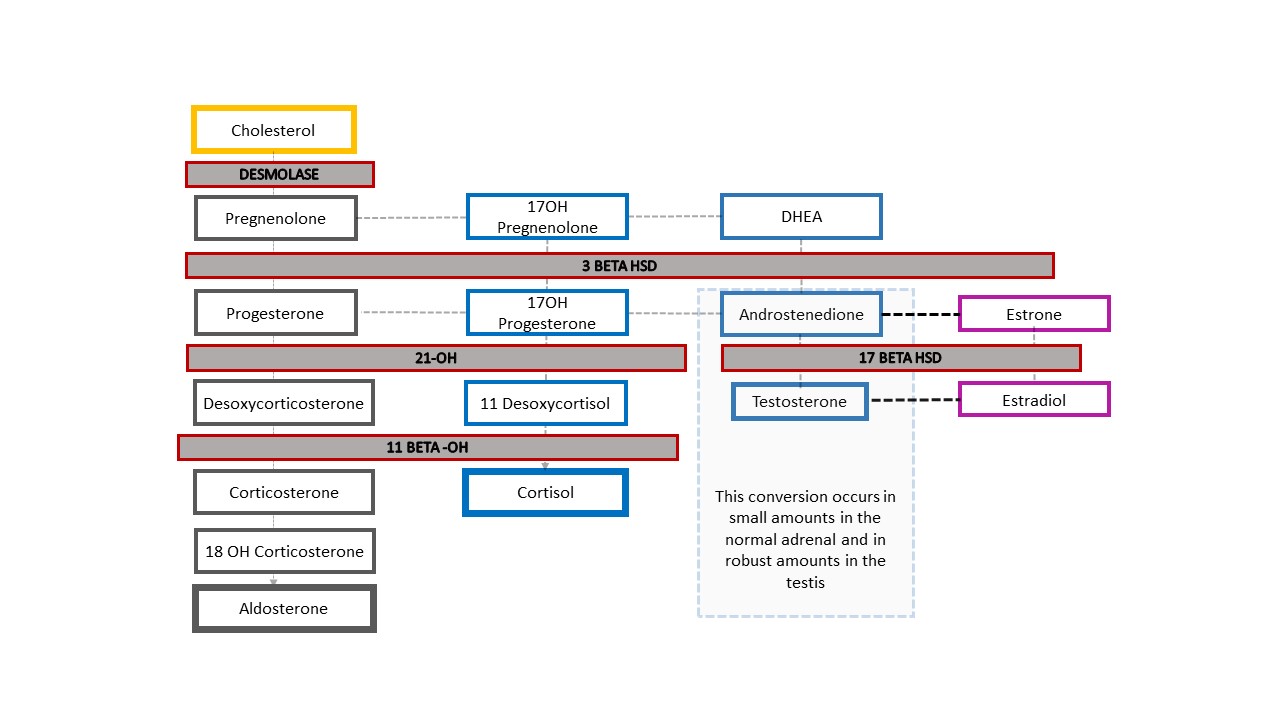
图 4 甾体合成通路及 CAH 诊断谱。 某一酶的生成存在任何程度的缺陷,都会导致下游产物减少,并使该通路分流至性类固醇方向。下丘脑-垂体-肾上腺轴受到刺激以进行代偿。ACTH 升高导致肾上腺皮质增生,并进一步增加前体的生成。
46,XX CAH 患者的典型解剖学表现包括不同程度的阴蒂增大和尿生殖窦形成。性腺为卵巢,且不可触及。存在多种突变,会导致具有不同特征的教科书式肾上腺类固醇内环境以及体征与生理表型。最常见的类型是21-羟化酶缺乏症。46,XX CAH 的婴儿可在生后约1周出现危及生命的失盐,因此对所有外生殖器不明确的婴儿均应考虑该诊断。17-羟孕酮升高具有诊断意义。女性性别认同以及按女性抚养最为常见,但并非绝对。
46,XY 性发育障碍
在46,XY DSD类别中,存在一系列性腺发育以及雄激素合成与作用方面的障碍,详见表3。与上文所述的AIS谱系情况相似,完全或部分性腺发育不全也可导致类似的解剖学变异:具有46,XY染色体的婴儿可呈现典型女性或不明确的表型(例如:近端尿道下裂并伴阴茎阴囊转位,以及一侧或双侧性腺未降)。完全型AIS和完全性腺发育不全的个体通常作为女性抚养,并自我认同为女性;而任一疾病的部分型患者在抚养性别和性别认同方面具有更大的变异性。
另一种 46,XY 性发育障碍——5α-还原酶缺乏症——会导致睾酮向二氢睾酮(DHT)的转化减少,进而出现多样的外生殖器表型。图 5 和 图 6 比较了典型的睾丸发育通路与发生于5α-还原酶缺乏症患者的通路。
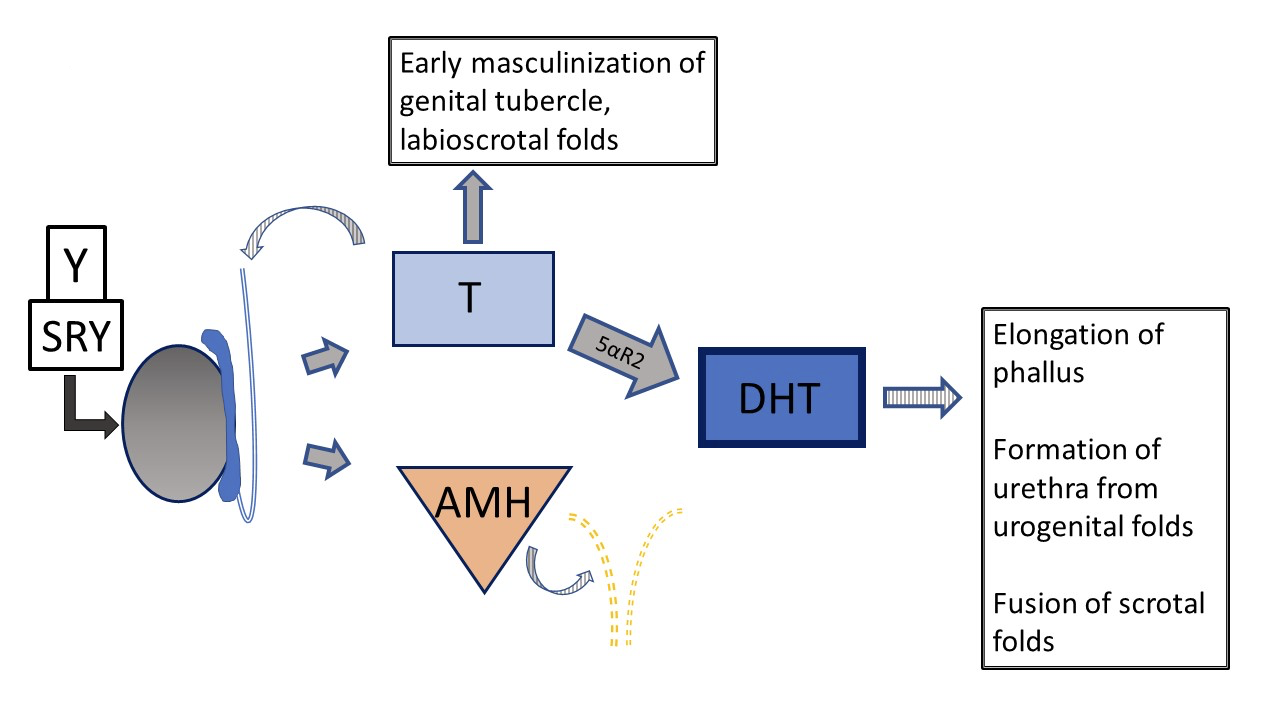
图 5 典型的46XY产前外生殖器发育 发育可以在多个不同阶段偏离“典型”。条纹箭头表示旁分泌样功能。虚线表示苗勒管在发育早期的退化。
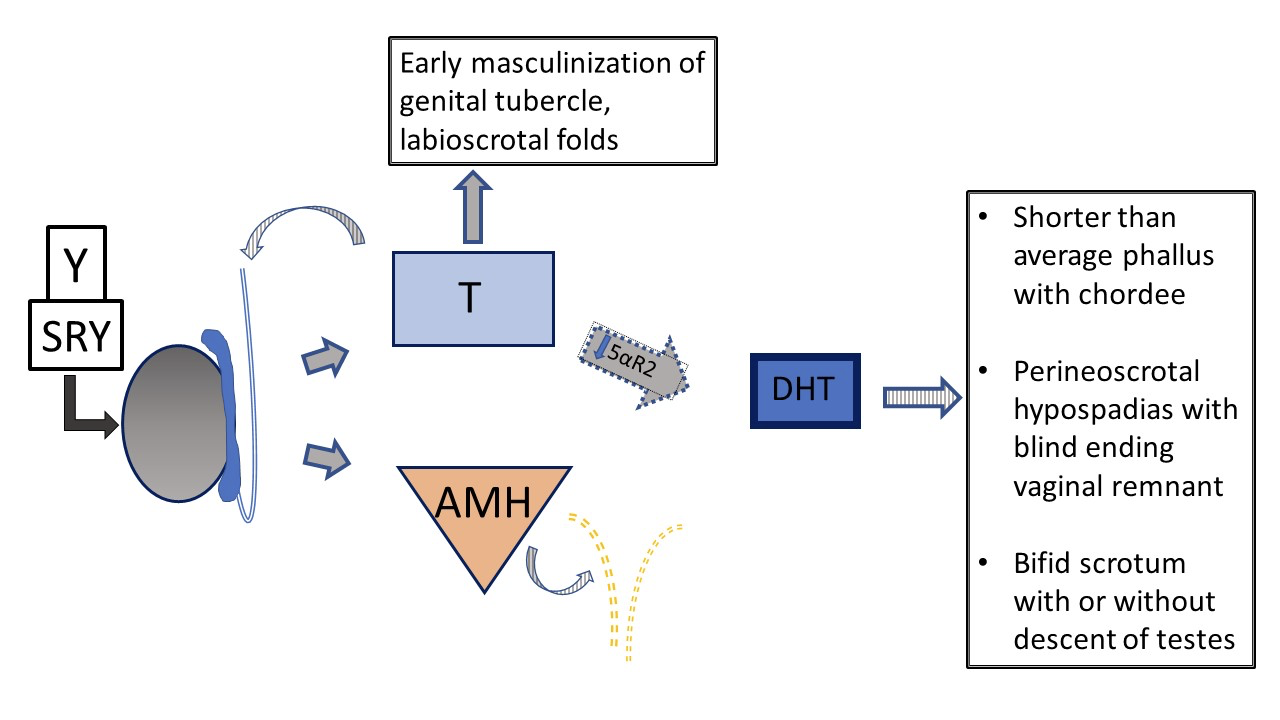
图 6 46XY 胎儿期外生殖器发育,见于患有 vs 5-α-还原酶缺乏的患者。睾丸已分化,且睾酮与 AMH 分泌正常。无子宫。由于 5-α-还原酶功能受损,T /DHT 比值显著升高。在 DHT 较低的情况下,表型具有变异性,从典型女性到典型男性,亦可为不明确的外生殖器解剖。若缺乏严重,阴茎与泌尿生殖窦的发育在男性化的早期阶段即停滞,会阴部仅有单一的泌尿生殖外口,且阴唇阴囊褶皱不融合。
图7 显示了在一名临床表现为外生殖器不明确的46,XY CAH患者这一罕见情形中所见的相应肾上腺甾体合成级联:3-β-羟类固醇脱氢酶[HSD]缺乏症。该酶的阻断发生在肾上腺甾体合成级联中脱氢表雄酮(DHEA)向雄烯二酮转化之前,因此婴儿会出现盐皮质激素和糖皮质激素减少,并伴有生殖器发育差异。与46,XX型CAH且为21-羟化酶或11β-羟化酶缺乏的婴儿类似,3-β-HSD缺乏的婴儿可出现危及生命的盐丢失。诊断依据为17-OH孕烯醇酮和DHEA升高。
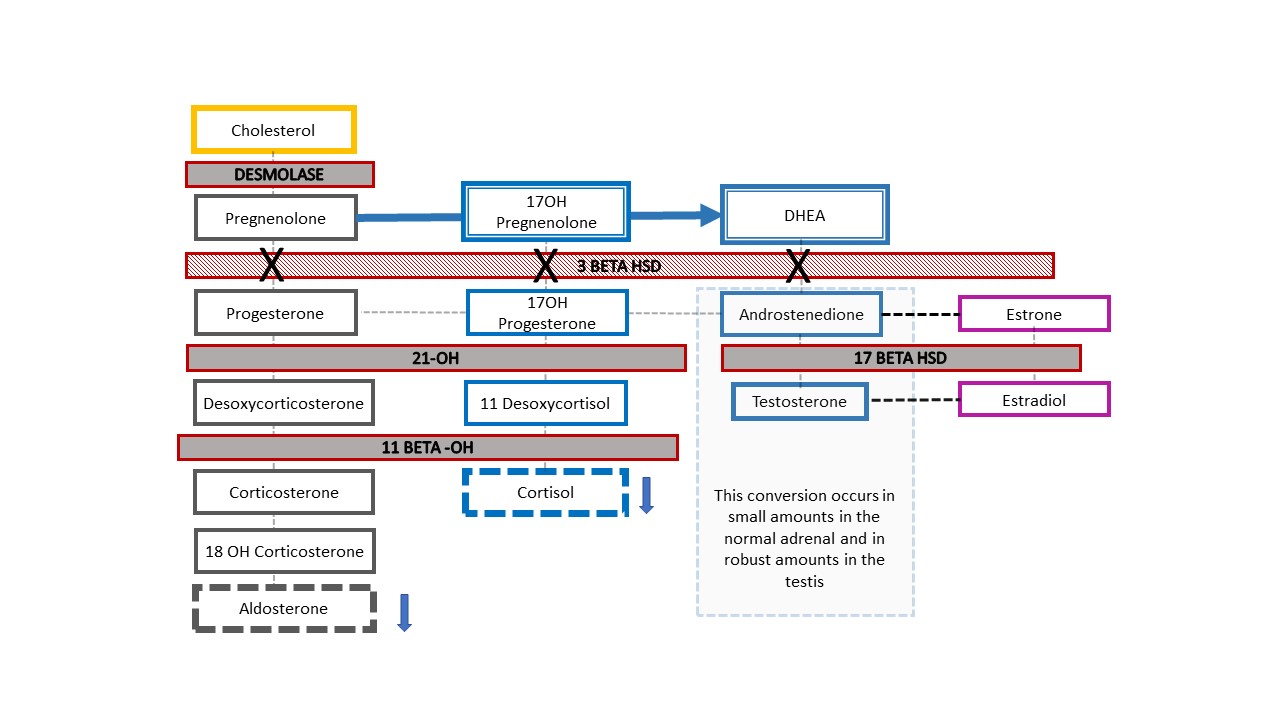
图 7 3-β-羟类固醇脱氢酶缺乏的类固醇生成通路。 肾上腺胆固醇级联中的这一相对近端缺陷会导致严重的糖皮质激素和盐皮质激素缺乏。其独特之处在于,患有CAH的46,XY婴儿外生殖器男性化不足,而46,XX婴儿则呈现典型(或近乎典型)女性的外生殖器外观。当一名在产前检测到Y染色体的新生儿出生时外生殖器外观不明确,这是需要排除的CAH类型。升高的17-羟孕烯醇酮和DHEA有助于确诊。
性染色体型DSD
在表 3中列出的性染色体DSD诊断中,具有45,X/46,XY(或类似)嵌合体核型的情况值得特别一提。具有此核型的患者可表现为女性型外生殖器(伴Y染色体成分的特纳综合征[TS+Y])、不明确的外生殖器(混合性性腺发育不良[MGD]),或男性型外生殖器。TS+Y患者的性腺肿瘤风险升高,这与45,X特纳综合征患者不同。MGD的表型以及抚养性别/性别认同具有多样性,然而常见的结果是性腺和阴唇阴囊不对称,如图 8所述。任何具有45,X/46,XY核型的患者均可能出现TS的表现(例如主动脉缩窄、肾脏异常),因此应接受与经典TS患者相同的系统性筛查检查。7,8
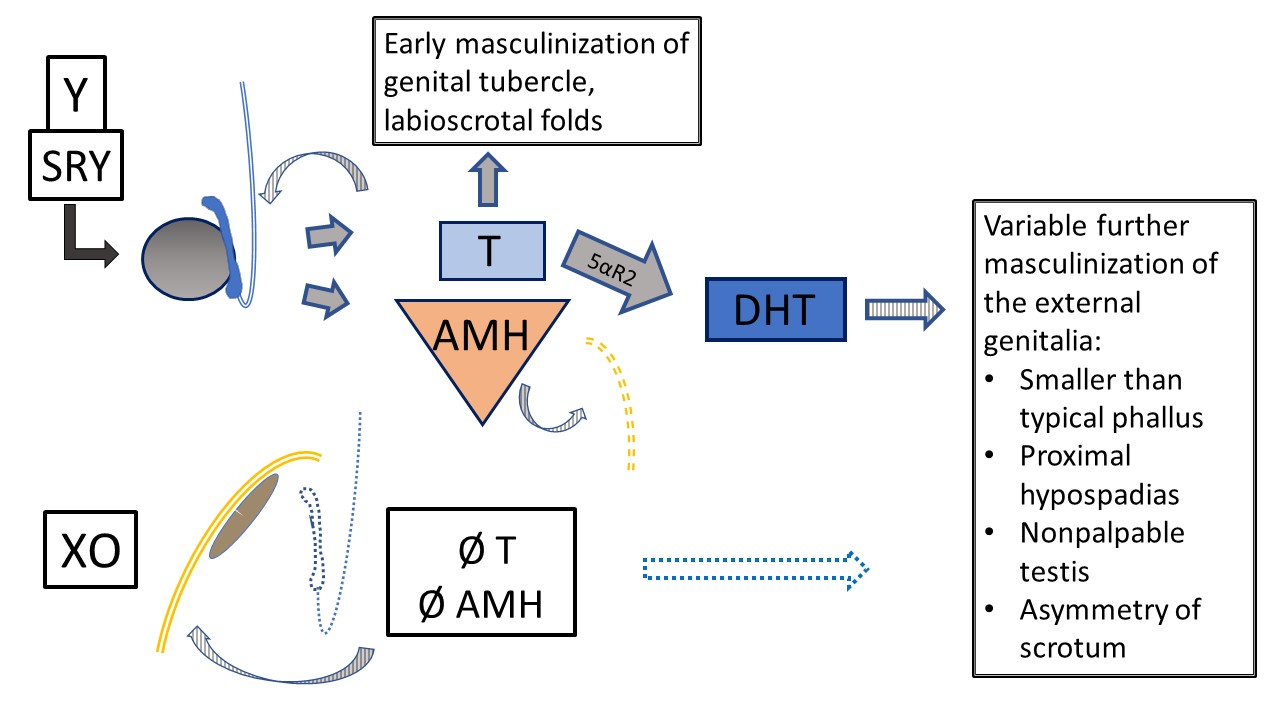
图 8 45,X/46,XY 混合性性腺发育不良。 这种嵌合核型可出现多种表型。该图示表示在一侧为睾丸(由 Y 染色体决定)、一侧为条索状性腺(默认发育,卵巢样间质)的情况下,内外生殖器的发育。经典表型(当观察到一定程度的外生殖器不明确时)为:一侧不可触及的性腺、一侧已下降的睾丸,以及近端尿道下裂。由于 T 和 AMH 具有旁分泌功能,条索状性腺常伴同侧半子宫和输卵管,且无沃尔夫管结构。由于存在 Y 物质,存在发生性腺母细胞瘤和无性细胞瘤的风险。睾丸发育不良程度不一,影响早期和远期功能及肿瘤风险。原发男性化发生,但尿道和阴囊的进一步发育受睾丸功能影响,程度不一。
卵睾性性发育障碍
卵睾性 DSD 指同一名个体同时存在卵巢和睾丸组织(旧称 – 真两性畸形)。通常,卵巢组织发育更好,而睾丸组织为发育不良。表型、养育性别/性别认同以及生育潜能均可有所不同。重要的是,卵睾性 DSD 患者可具有任何核型(尽管 46, XX 最常见),因此对任何以不典型外生殖器就诊的患者都必须考虑这一诊断。
多学科评估
除了提出不那么带有贬义的命名法之外,2006 年共识声明的其他基石属于照护范式,包括由多学科团队进行的咨询,以及对患者及其父母的纵向心理社会支持。1
多学科团队
在新生儿住院会诊中与家庭合作的团队,最好与随后为患者及其家庭提供纵向(连续性)照护的团队为同一团队。该团队负责向家庭讲解疾病情况及所有潜在的管理选项;在需要作出决定之处(例如,抚养性别、信息披露、手术)提供支持,并与家庭开展共同决策。理想情况下,团队应包括以下成员:
- 内分泌科
- 心理健康 (心理学家、社会工作者)
- 泌尿外科 / 小儿外科 / 妇科
- 遗传咨询师 / 医学遗传科医师
- 护理
- 患者倡导者 / 患者教育者
- 新生儿科医师 (住院会诊)
每位专业人员都为多学科团队带来不同的视角、相关的临床专长以及人际沟通能力。尽管早期或紧急手术的需求并不常见,外科医生仍然是团队中重要的一员。所有成员都参与团队讨论,并为患者及其家庭的就医体验作出贡献。理想情况下,彼此尊重并愿意兼顾各方观点能够增强团队运作,使团队随着时间推移提供更高水平的照护,尤其是在诊疗标准不断演变的过程中。
每一位患有 DSD 状况的个体可能无法获得功能完善的多学科团队支持。有些人可能不愿意与团队合作,或认为长期随访负担较重。因此,每一次关于诊断及潜在长期问题或医疗需求的早期宣教机会都非常重要。在长期随访可行且可接受的情况下,这些工作可在更为常规的单一专科照护框架内完成,视患者情况而定。例如,在作者所在机构,一些 CAH 患者每季度与其内分泌科医师随访,并每年参加一次完整的多学科团队联合门诊。
积极协作,彼此学习并向患者/家庭学习,有助于团队运转并推动照护进步。团队借鉴既往经验并吸收新知识,尤其是在患者体验和下一代遗传学检测领域。团队还会识别知识层面以及照护范式中的空白,这些空白可通过协作与有目的的研究加以解决。定期开展以病例或专题为基础的多学科会议,有助于团队成员和学习者在各类病情方面建立熟练度,并提供机会理解理念上的差异。
对疑似性发育障碍(DSD)患者的初始处理
疑似 DSD 的患者典型地在婴儿期因不典型外生殖器就诊,或在青春期因原发性闭经就诊。DSD 情况也可能因影像学偶然发现或术中发现、女孩出现腹股沟疝,或女孩在青春期出现男性化而进入医疗视野。越来越多地,DSD 在产前即被怀疑,原因是无细胞 DNA 与超声检查结果不一致,或超声提示外生殖器发育存在差异。
当一名疑似 DSD 的新生儿出生时,建议在可行的情况下会诊多学科团队(如上所述)。请记得向家庭祝贺他们迎来新生儿(这一步常被忽视,尤其是在对 DSD 感到意外或不熟悉时)。如果对性别指定有任何疑问,在提及婴儿时使用性别中立的用语(用“你们的宝宝”,而不是“它”)。图 9 展示了描述新生儿外生殖器检查的推荐用语,包括可使用的性别中立术语。除详细的产前史、家族史和体格检查外,针对疑似 DSD 患者的初始评估建议包括:
- 核型
- 内分泌检查 (17-羟孕酮, 睾酮, 黄体生成素, 促卵泡激素, 电解质)
- 骨盆超声 以评估性腺的位置和类型,以及是否存在苗勒氏管结构
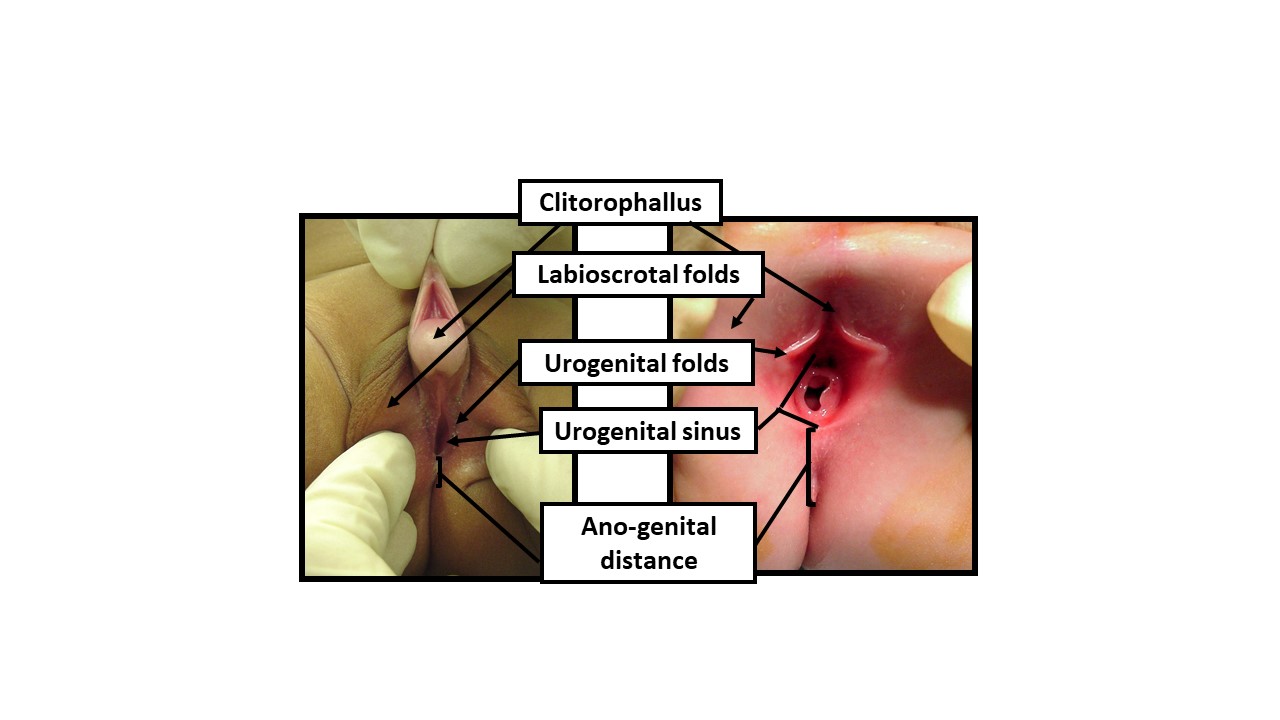
图 9 使用性别中立的解剖学术语描述新生儿体检。 在检查外生殖器外观不明确的新生儿时,应使用性别中立术语,直至有足够资料允许与父母共同决策以确定养育性别。体检应记录以下反映雄激素暴露的指标: 1) 阴蒂-阴茎结构的长度, 2) 龟头宽度, 3) 会阴开口的数目和位置, 4) 阴唇-阴囊褶皱的融合程度, 5) 阴唇-阴囊褶皱的色素沉着过度和皱褶, 6) 肛生殖距离, 7) 任何可触及的性腺的位置
采用基于可触及性腺数目以及上述内分泌、遗传学和影像学结果的算法化评估,可有效缩小鉴别诊断范围。在新生儿期最为关键的是排除危及生命的盐丢失型先天性肾上腺皮质增生症(CAH)。上述多学科团队在这一关键的诊断阶段均发挥作用,帮助家庭整合新的信息,并就抚养性别、信息披露以及手术作出决策。
手术治疗选项
针对性发育差异(DSD)患者的手术选择高度个体化,理想情况下,手术治疗的决策应在多学科团队的框架内做出。根据患者的需求和目标,手术可以是诊断性的、重建性的,或切除性的。
诊断检查
当在完成内分泌与遗传学评估以及详细的影像学检查之后,解剖结构或诊断仍不明确时,通常会进行诊断性操作。
内镜检查可对泌尿生殖道进行详细的可视化观察和测量。例如,可界定泌尿生殖窦的长度和构型。阴道结构可评估其深度、数目,以及是否存在宫颈。
诊断性腹腔镜检查有助于观察性腺和苗勒管结构。这种可视化有助于诊断,并为未来的重建手术规划提供信息。性腺活检可通过腹腔镜或开放手术入路进行,以确定组织类型(例如,若怀疑卵睾型 DSD),或在怀疑存在 Y 染色体物质但外周血核型分析尚未检出的情况下将标本送检核型分析。图 10 显示了一名 MGD 患者的腹腔镜静态图像:临床上曾怀疑 MGD,并由诊断性腹腔镜和活检所提供的性腺大体及组织学外观予以证实。
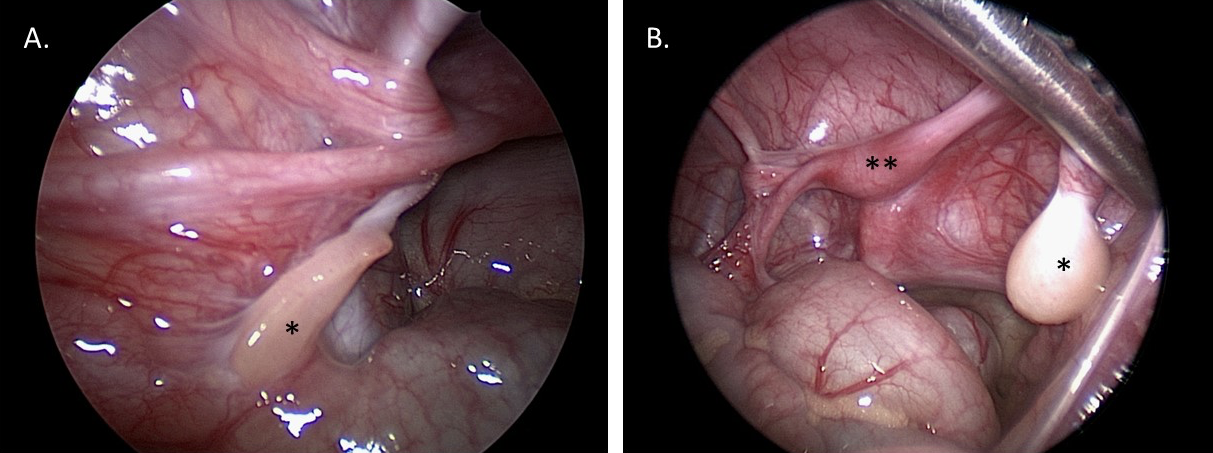
图 10 1 岁混合性性腺发育不良患儿骨盆的诊断性腹腔镜图像。A. *左侧条索状性腺。B. *右侧发育不良睾丸。 **子宫。
男性化生殖器成形术
对于DSD患者,男性化生殖器成形术最常涉及近端尿道下裂修复术,且常需同时行阴囊成形术以矫正阴茎阴囊转位和/或阴囊裂。尿道下裂修复术的技术细节与无明确DSD诊断的患者相似。在DSD患者中,尿道下裂修复术通常需要两期手术,以矫正阴茎下弯并建立终末性尿道外口。关于已知DSD诊断的患者与无特定DSD的近端尿道下裂患者相比,其尿道下裂手术结局是否更差,文献中仍存在争议。9,10,11
女性化生殖器成形术
组成部分包括阴蒂成形术(伴阴唇成形术)和阴道成形术。存在多种手术方法,可根据患者的解剖结构和目标进行个体化定制。一名先天性肾上腺增生(CAH)患者接受部分尿生殖道动员术(PUM)及阴唇成形术而未进行阴蒂成形术,其术前手术标记示例如图11所示。
阴蒂成形术与阴唇成形术
最常见报道的现代阴蒂成形术包括保留神经的阴蒂缩小术。12 手术通过对阴蒂进行去套式剥离并切除海绵体(勃起)组织,同时避开背侧神经血管束。为进一步保护阴蒂头的血供和感觉,引入了保留白膜的缩小性阴蒂成形术,其做法是在腹侧对海绵体切开并切除勃起组织,同时保留周围白膜。阴蒂头常予保留,近端回缩并固定于耻骨下方。阴蒂头缩小可通过切除腹侧楔形组织并将腹侧切缘重新对合来完成;但该方法存在降低阴蒂头感觉的风险。通过阴蒂成形术联合阴唇成形术,常可达到阴蒂隐匿的外观,因此阴蒂头缩小术的实施越来越少见。
作为上述阴蒂缩小术的替代方案,已经出现了若干种保留海绵体的技术。例如,Pippi Salle 描述了一种海绵体分离术。13 在该术式中,双侧阴蒂海绵体与阴蒂头及其神经血管束完全分离,且彼此之间也相互分离。随后,将每侧海绵体分别经隧道置入相应的同侧大阴唇内,并将阴蒂头向近端重新定位,类似于阴蒂缩小术。该 ‘Pippi Salle 术式’ 具有理论上的可逆性优势,因为未切除任何组织,然而关于逆转的尝试及其他远期结局尚未有报道。
阴唇成形术通常是阴蒂成形术(可伴或不伴阴道成形术)手术的最后一步。其目标是将阴蒂包皮和阴蒂体的组织在阴蒂表面重新配置,以遮盖阴蒂头,并将皮肤边缘向后推进,使其位于阴道前庭/阴道口的外侧。
阴道成形术
阴道成形术的处理方式涉及评估患者是否存在低位汇合的尿生殖窦(共同通道较短,阴道与尿道的分离点靠近会阴)或高位汇合(共同通道较长,分离位置更高。14
会阴皮瓣阴道成形术(Fortunoff 技术)对于低位汇合型尿生殖窦的患者可能已足够。对于该技术,制作一个后方U形皮瓣(如图11所示)。随后在发育不良的汇合处的近端切开阴道后壁,并将该皮瓣置入并固定于该阴道切口内,以增大阴道腔径。
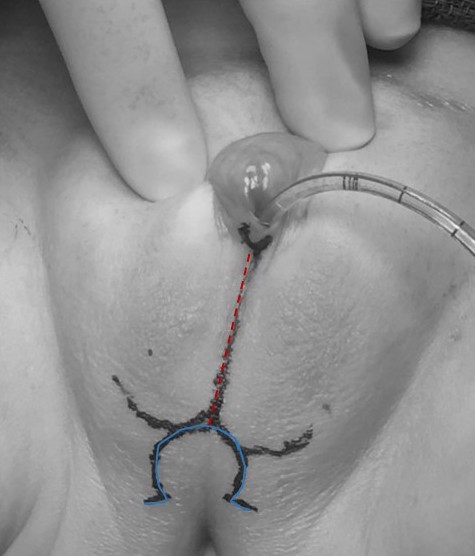
图 11 对一名先天性肾上腺皮质增生患者的术前手术标记:计划进行部分泌尿生殖窦游离术和阴唇成形术,且不行阴蒂成形术。 在泌尿生殖窦开口内可见一根导尿管。正中线标记(红色虚线)延伸穿过融合的阴唇阴囊褶,Ω 形标记(蓝色实线)将形成用于阴道成形术的后方皮瓣。
在整体泌尿生殖道动员(TUM)或部分泌尿生殖道动员(PUM)中,将尿道与阴道作为一个整体向会阴方向动员。15 二者的区别在于,是否在耻骨后进行的前向分离中处理(切断)耻骨尿道韧带,如图 12所示。更广泛的动员可能带来尿失禁的隐患,但尚未被明确证实;若汇合部至膀胱颈的距离缩短,则更值得担忧。用于低位汇合病例时,PUM通常可将阴道带至可予外置,或可用以后方为蒂的会阴皮瓣进行桥接的范围(见上文)。高位汇合提示解剖更为复杂,包括达到会阴的距离更长、阴道与膀胱颈关系更近,且常伴阴道较短。直接拉出术从长期看未必有利。在高位汇合病例中,即便进行泌尿生殖道动员,仍需将阴道自泌尿生殖窦分离并拉出至会阴。在此情况下,泌尿生殖窦成为尿道。阴道可借助会阴皮瓣,或将多余的已动员的泌尿生殖窦组织制成皮瓣,以到达会阴。
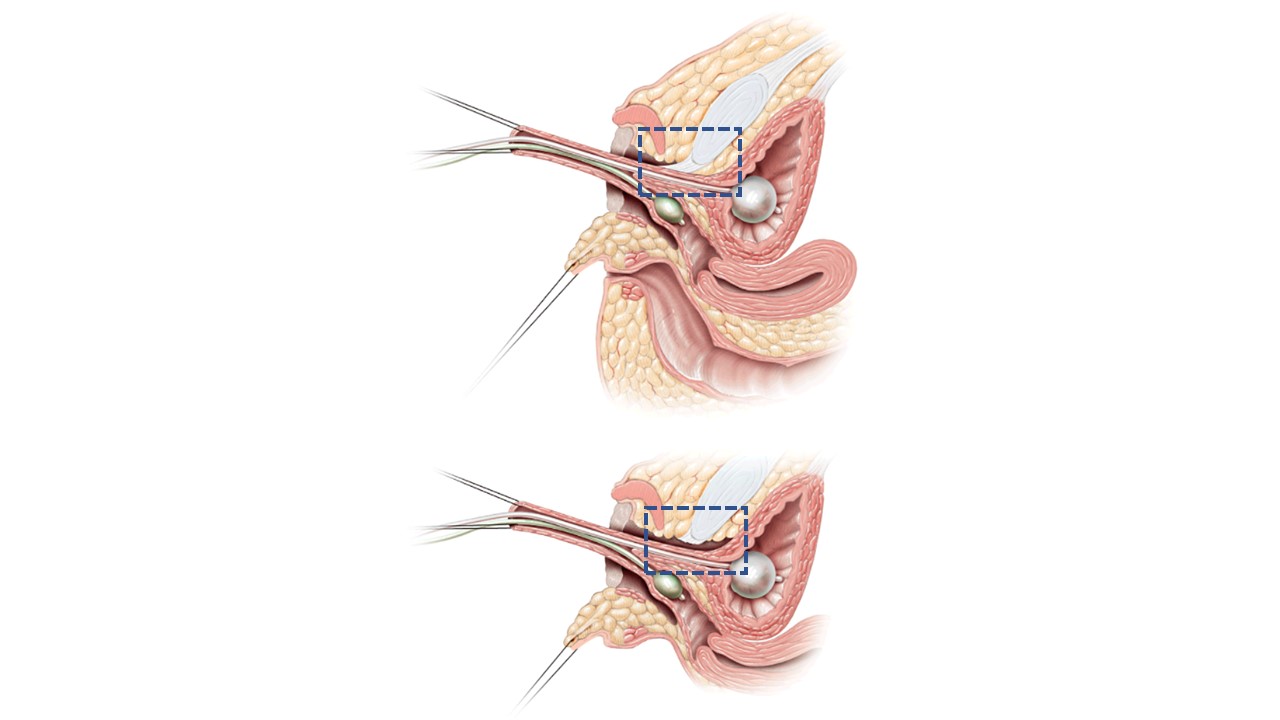
图 12 部分与完全尿生殖动员的矢状位断面图像。A. 部分尿生殖动员:尿道与阴道作为一个整体向会阴前移。耻骨尿道韧带(蓝色虚线框)保留完整。B. 完全尿生殖动员:尿道与阴道作为一个整体向会阴前移。耻骨尿道韧带(蓝色虚线框)被切断。改编自:Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int 2008; 102 (9): 1182–1197. DOI: 10.1111/j.1464-410x.2008.08091.x. (于 2022/2/1 获准)
对于先天缺如或发育不良的苗勒管结构患者,阴道成形术包括通过钝性分离在膀胱与直肠之间的膀胱后间隙内建立新阴道,然后以自体组织(例如,皮肤移植、颊黏膜移植、腹膜瓣)或其他生物移植物(例如,小肠黏膜下层 [SIS])铺衬这一新的潜在腔隙。这些技术也可作为对上述手术的辅助,或用于再手术情境。
阴道扩张
一些患有DSD(例如,完全型AIS)的患者,通过定期进行阴道扩张,可以形成足以使性活动舒适且愉悦的阴道,而无需进行外科阴道成形术。所有接受外科阴道成形术的患者在术后恢复愈合期间也需要进行阴道扩张,并持续至其能够规律地进行阴道性交为止。
性腺切除术与性腺管理
针对 DSD 患者的性腺管理范式已随时间演变。传统上,患有 DSD 且肿瘤形成风险增加的患者被建议接受性腺切除术。通常假定其不育。随着时间推移,已明确肿瘤风险存在广泛差异,且所形成的肿瘤多为性腺母细胞瘤(非恶性)。16,17 无性细胞瘤(恶性生殖细胞肿瘤)也可能出现,但在青春期前极为罕见。对于先前被认为不育的部分人群,亦可能存在非常规的生育潜能。18 因此,性腺管理建议正日益个体化,需考虑患者的诊断(从而可预测的肿瘤风险)、年龄,以及内源性性腺功能(包括激素与生殖功能)的潜力。对于选择行性腺切除术的患者,性腺组织冷冻保存是一种新兴选项,在假定未来辅助生殖技术按预期取得进展的前提下,可能允许获得生物学生育力。19 对于选择保留性腺的患者,目前尚无循证的监测方案;作者所在机构通常建议每 6–12 个月行一次盆腔超声检查。表4 显示了若干 DSD 诊断的性腺处理建议策略。
表 4 按诊断划分的性腺管理方案。 * 考虑实验性的性腺组织冷冻保存。
| 诊断 | 估计肿瘤风险 | 预期有生殖细胞? | 预期有青春期? | 性腺管理方案 |
|---|---|---|---|---|
| 46, XY 完全性性腺发育不全(Swyer 综合征) | 高 – 最高可达 35% |
否 (可能有生殖细胞前体) |
否 | -性腺切除术* |
| 部分性雄激素不敏感综合征 | 高 – 最高可达 50%,取决于性腺位置 |
是 可能有精子 |
是,差异较大 | -睾丸固定术,考虑活检 -超声随访观察 -性腺切除术* |
| 特纳综合征 + Y 染色体 | 中等 – 约 10-30% |
可能 – 可能有卵子 | 可能 | -超声随访观察 -性腺切除术*(标准做法,证据等级低) |
| 完全性雄激素不敏感综合征 | 低 – 约 2-10% |
是 – 精子前体 |
是 – 由于睾酮芳香化为雌激素 | -青春期后性腺切除术* -超声、MRI 随访观察 |
苗勒管结构的处理
对于被作为男性抚养、且存在持续性苗勒管结构(即较大的前列腺小囊)的患者,通常予以原位保留,除非出现症状。切除存在潜在损伤输精管结构(以及可能或涉及盆腔神经)的风险,而患者往往无症状。尿道下裂修复是安全的,且即便存在较大的前列腺小囊,也不要求在尿道下裂修复前先行切除。若出现感染或令人困扰的周期性血尿等症状,则可经开放、腹腔镜或机器人手术途径行切除。对于因苗勒管残余梗阻而出现周期性疼痛,或令人困扰的周期性血尿的患者,也可考虑采用以月经抑制为目的的激素治疗。
结论
性发育差异(DSD)涵盖了由染色体、性腺或解剖发育异常所致的广泛疾病与状况。即便在特定的 DSD 诊断与类别内,也存在广泛的表型差异。在可能的情况下,建议采用多学科协作的评估与长期随访。
要点
- 医学上通用的 DSD 术语并非所有受影响的个体及其家属都偏好使用。在具体的临床交流中,询问患者及家属在提及诊断和解剖时更愿意使用哪些术语。
- DSD 状况源于非典型的染色体、性腺或激素作用。思考异常所在的层面,并判定所识别的缺陷是完全性还是部分性,有助于预测患者表型。
- 可触及性腺的数量构成 DSD 鉴别诊断的基础。随后通过核型、内分泌实验室检查和盆腔超声进一步细化鉴别。
- 记得向有疑似 DSD 新生儿的新手父母表示祝贺——这一步至关重要,却常被忽视。
- CAH 是 46, XX DSD 最常见的形式。临床特征包括男性化的外生殖器且性腺不可触及。危及生命的盐丢失可在生后 1 周出现。
- 卵睾性 DSD 可发生于任何核型的患者。
- 45, X/46, XY 核型的患者可具有女性型、模糊或男性型的外生殖器。任何具有该核型的患者都可能出现 Turner 综合征的全身性表现。
推荐阅读
- Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
- Kaefer M, Diamond D, Hendren WH. The Incidence Of Intersexuality In Children With Cryptorchidism And Hypospadias: Stratification Based On Gonadal Palpability And Meatal Position. J Urol 1999; 162 (3 Part 2): 1006–1007. DOI: 10.1016/s0022-5347(01)68049-2.
- Kim S, Rosoklija I, Johnson EK. Surgical, Patient, and Parental Considerations in the Management of Children with Differences of Sex Development. Curr Pediatr Rep 2018; 6 (3): 209–219. DOI: 10.1007/s40124-018-0177-4.
- Mouriquand PD, Gorduza DB, Gay CL. Re: “Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how?” J Pediatr Urol 2016; 12 (6): 442–443. DOI: 10.1016/j.jpurol.2016.07.013.
- Nixon R, Cerqueira V, Kyriakou A, Lucas-Herald A, McNeilly J, McMillan M, et al.. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod 2017; 32 (10): 2130–2137. DOI: 10.1093/humrep/dex280.
- Van Batavia JP, Kolon TF. Fertility in disorders of sex development: A review. J Pediatr Urol 2016; 12 (6): 418–425. DOI: 10.1016/j.jpurol.2016.09.015.
参考文献
- Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
- Johnson EK, Rosoklija I, Finlayson C. Faculty Opinions recommendation of Attitudes towards "disorders of sex development" nomenclature among affected individuals. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2017; 13: 608. DOI: 10.3410/f.727655647.793533819.
- D’Oro A, Rosoklija I, Jacobson DL, Finlayson C, Chen D, Tu DD, et al.. Patient and Caregiver Attitudes toward Disorders of Sex Development Nomenclature. J Urol 2020; 204 (4): 835–842. DOI: 10.1097/ju.0000000000001076.
- Davies JH, Knight EJ, Savage A, Brown J, Malone PS. Evaluation of terminology used to describe disorders of sex development. J Pediatr Urol 2011; 7 (4): 412–415. DOI: 10.1016/j.jpurol.2010.07.004.
- Lin-Su K, Lekarev O, Poppas DP, Vogiatzi MG. Congenital adrenal hyperplasia patient perception of ‘disorders of sex development’ nomenclature. Int J Pediatr Endocrinol 2015; 2015 (1): 9. DOI: 10.1186/s13633-015-0004-4.
- Snodgrass W, Macedo A, Hoebeke P, Mouriquand PDE. Hypospadias dilemmas: A round table. J Pediatr Urol 2011; 7 (2): 145–157. DOI: 10.1016/j.jpurol.2010.11.009.
- Wu Q, Wang C, Shi H, Kong X, Ren S, Jiang M. The Clinical Manifestation and Genetic Evaluation in Patients with 45,X/46,XY Mosaicism. Sex Dev 2017; 11 (2): 64–69. DOI: 10.1159/000455260.
- Das DV, Jabbar PK. Clinical and Reproductive Characteristics of Patients with Mixed Gonadal Dysgenesis (45,X/46, XY). J Obstet Gynaecol India 2021; 71 (4): 399–405. DOI: 10.1007/s13224-021-01448-3.
- Saltzman AF, Carrasco A, Colvin A, Campbell JB, Vemulakonda VM, Wilcox D. Patients with disorders of sex development and proximal hypospadias are at high risk for reoperation. World J Urol 2018; 36 (12): 2051–2058. DOI: 10.1007/s00345-018-2350-3.
- Ochi T, Ishiyama A, Yazaki Y, Murakami H, Takeda M, Seo S, et al.. Surgical management of hypospadias in cases with concomitant disorders of sex development. Pediatr Surg Int 2019; 35 (5): 611–617. DOI: 10.1007/s00383-019-04457-6.
- Palmer BW, Reiner W, Kropp BP. Proximal Hypospadias Repair Outcomes in Patients with a Specific Disorder of Sexual Development Diagnosis. Adv Urol 2012; 2012: 1–4. DOI: 10.1155/2012/708301.
- Kaefer M, Rink RC. Treatment of the Enlarged Clitoris. Front Pediatr 2017; 5. DOI: 10.3389/fped.2017.00125.
- Pippi Salle JL, Braga LP, Macedo N, Rosito N, Bagli D. Corporeal Sparing Dismembered Clitoroplasty: An Alternative Technique for Feminizing Genitoplasty. J Urol 2007; 178 (4s): 1796–1801. DOI: 10.1016/j.juro.2007.03.167.
- Guarino N, Scommegna S, Majore S, Rapone AM, Ungaro L, Morrone A, et al.. Vaginoplasty for Disorders of Sex Development. Front Endocrinol (Lausanne) 2013; 4: 29, DOI: 10.3389/fendo.2013.00029.
- Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int 2008; 102 (9): 1182–1197. DOI: 10.1111/j.1464-410x.2008.08091.x.
- Looijenga LHJ, Hersmus R, Oosterhuis JW, Cools M, Drop SLS, Wolffenbuttel KP. Tumor risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab 2007; 21 (3): 480–495. DOI: 10.1016/j.beem.2007.05.001.
- Looijenga LHJ, Hersmus R, Leeuw BHCGM de, Stoop H, Cools M, Oosterhuis JW, et al.. Gonadal tumours and DSD. Best Pract Res Clin Endocrinol Metab 2010; 24 (2): 291–310. DOI: 10.1016/j.beem.2009.10.002.
- Finlayson C, Fritsch MK, Johnson EK, Rosoklija I, Gosiengfiao Y, Yerkes E, et al.. Presence of Germ Cells in Disorders of Sex Development: Implications for Fertility Potential and Preservation. J Urol 2017; 197 (3 Part 2): 937–943. DOI: 10.1016/j.juro.2016.08.108.
- Harris CJ, Corkum KS, Finlayson C, Rowell EE, Laronda MM, Reimann MB, et al.. Establishing an Institutional Gonadal Tissue Cryopreservation Protocol for Patients with Differences of Sex Development. J Urol 2020; 204 (5): 1054–1061. DOI: 10.1097/ju.0000000000001128.
- Kaefer M, Diamond D, Hendren WH. The Incidence Of Intersexuality In Children With Cryptorchidism And Hypospadias: Stratification Based On Gonadal Palpability And Meatal Position. J Urol 1999; 162 (3 Part 2): 1006–1007. DOI: 10.1016/s0022-5347(01)68049-2.
- Kim S, Rosoklija I, Johnson EK. Surgical, Patient, and Parental Considerations in the Management of Children with Differences of Sex Development. Curr Pediatr Rep 2018; 6 (3): 209–219. DOI: 10.1007/s40124-018-0177-4.
- Mouriquand PD, Gorduza DB, Gay CL. Re: “Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how?” J Pediatr Urol 2016; 12 (6): 442–443. DOI: 10.1016/j.jpurol.2016.07.013.
- Nixon R, Cerqueira V, Kyriakou A, Lucas-Herald A, McNeilly J, McMillan M, et al.. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod 2017; 32 (10): 2130–2137. DOI: 10.1093/humrep/dex280.
- Van Batavia JP, Kolon TF. Fertility in disorders of sex development: A review. J Pediatr Urol 2016; 12 (6): 418–425. DOI: 10.1016/j.jpurol.2016.09.015.
最近更新时间: 2025-09-22 08:00