
13: 肾脏囊性疾病
阅读本章大约需要 10 分钟。
引言
肾囊性疾病包括一大类可为散发或遗传性、先天或后天获得性的肾脏疾病,其表现可为单侧或双侧肾脏内从单个囊肿到多个囊肿不等(见下方表 1中的遗传性分型)。1 依据1987年分类、命名与术语委员会的意见,肾囊性疾病的分类主要由是否为遗传性或非遗传性来决定。2 肾囊性疾病多可视为罕见病,但可能伴随严重后果,如慢性肾脏病(CKD)、肾功能衰竭或肝脏疾病。尽管总体罕见,常染色体显性多囊肾(ADPKD)却是人类最常见的遗传性疾病之一。儿科人群中肾囊性疾病可能出现严重并发症,尤其是CKD,因此早期诊断和有效治疗手段尤为重要。
表 1 肾囊性疾病的遗传性分类。1
| 遗传性 | 非遗传性 |
|---|---|
| 常染色体隐性(婴儿型)多囊肾病 | 多囊性肾(多囊性发育不良肾) |
| 常染色体显性(成人型)多囊肾病 | 良性多房性囊肿(囊性肾瘤) |
| 少年型肾单位发育不全与肾髓质囊性病复合征 | 单纯性囊肿 |
| 少年型肾单位发育不全(常染色体隐性) | 肾髓质海绵肾 |
| 肾髓质囊性病(常染色体显性) | 散发性肾小球囊性肾病 |
| 先天性肾病综合征(家族性肾病综合征)(常染色体隐性) | 获得性肾囊性病 |
| 家族性发育不全型肾小球囊性肾病(常染色体显性) | 肾盏憩室(肾盂源性囊肿) |
| 伴肾囊肿的多发畸形综合征(如结节性硬化症、冯·希佩尔-林道病) |
胚胎学
囊肿通常继发于在上皮细胞层面的分泌、吸收及电解质失衡,并可在肾小管系统内的任何部位形成。关于遗传性囊性肾病的最新研究提示,所涉及的相关基因与肾上皮细胞的初级纤毛有关,这可能是这些疾病的共同通路。3 术语“纤毛病”指的就是这一共同通路。纤毛的功能是调节肾小管细胞的增殖与分化,其功能障碍可能导致肾小管异常扩张并形成囊肿。4
本章将讨论各种遗传性和非遗传性囊性肾病的流行病学、发病机制、评估与治疗,包括常染色体显性多囊肾病(ADPKD)、常染色体隐性多囊肾病(ARPKD)、孤立性肾囊肿、肾盏憩室、多囊性发育不良肾(MCDK)、髓质海绵肾、肾单位发育不全、多房性囊肿(多房性囊性肾瘤)、获得性囊性肾病,以及与囊性肾病相关的综合征。
常染色体显性多囊肾病
常染色体显性多囊肾病(ADPKD)较为常见,其发生率约为每400至1,000个活产中有1例,临床表现为肾单位的囊性扩张以及其他肾外异常,如颅内动脉瘤。5 疾病严重程度因人而异,并倾向于随时间而加重,因此许多儿科患者,甚至部分中年成年人,可能仍无症状。尽管并不常见,但儿童期出现的重症可能预示显著的疾病相关并发症负担和死亡率。6 由于家族史或影像学的偶然发现,越来越多无症状的儿童被诊断为患有ADPKD。
流行病学
在ADPKD患者中,约有96%将在90岁时出现该病的临床表现。7 尽管多数病例在中年患者(30–40岁)中被发现,且40岁之前发生肾衰竭的情况罕见,但已有报道其可早至新生儿期即被描述。当见于新生儿时,ADPKD被认为更具侵袭性。8 尽管理论上为外显率100%的常染色体显性遗传模式,仍有10%的病例可能为散发发生。1
发病机制
ADPKD 以常染色体显性方式遗传,主要的基因突变分别发生在16号染色体上的PKD1和4号染色体上的PKD2。5 这些基因的产物是跨膜蛋白(分别为多囊蛋白1和多囊蛋白2),定位于初级纤毛内。9 位于16号染色体的PKD1突变占多数病例并与疾病严重程度更高相关,而其余大多数病例来自PKD2突变。10
多囊蛋白1或多囊蛋白2的功能异常会导致cAMP、ERK和mTOR等增殖通路信号的失调。纤毛可能在这些信号的传导中发挥组织协调作用,因为这些信号传导的失调会导致囊肿形成。11
评估与诊断
作为一种具成本效益且微创的影像学检查,肾脏超声常用于发现 ADPKD。30 岁以下者的超声诊断标准包括单侧或双侧至少存在两个囊肿;随着年龄增加,提示诊断所需的囊肿数量更多。12 然而,在儿科人群中,ADPKD 与 ARPKD 可能难以区分,因此阳性家族史的存在对诊断至关重要。13 在缺乏家族史的情况下,可进行基因检测。当家族史为阴性时,还需考虑的其他重要因素包括双侧肾脏增大、三个或以上的肝囊肿、脑动脉瘤,以及其他器官(蛛网膜、松果体、胰腺或脾)的孤立性囊肿。1,14 肝脏影像或活检也有助于鉴别 ADPKD 与 ARPKD,因为 ARPKD 始终存在肝纤维化,而 ADPKD 罕见。5 最后,肾脏病理(活检)亦可提供帮助,因为 ADPKD 可累及整个肾小管,甚至包括肾小球,然而 ARPKD 不会出现肾小球囊肿。5
治疗方案、预后及并发症
目前尚无治愈ADPKD的方法。治疗目标包括延缓终末期肾病(ESRD)的发生,并减少与肾功能下降、心脏疾病和颅内出血相关的ADPKD并发症负担。并发症受高血压管理的影响最为显著。1 因此,建议严格控制血压,血管紧张素转换酶(ACE)抑制剂和血管紧张素受体阻滞剂是常用选择,但关于理想的降压药尚无共识。15,16
正在研究中的新兴疗法旨在通过使用mTOR抑制剂、生长抑素、酪氨酸激酶抑制剂、Src激酶抑制剂、MEK抑制剂以及细胞周期蛋白依赖性激酶抑制剂来降低cAMP,从而限制囊肿的生长。
常染色体隐性多囊肾病
ARPKD 的特征是在婴儿中由于集合管系统内囊肿形成而导致双肾迅速增大。它可能与先天性肝纤维化相关,后者可导致门静脉高压。ARPKD 的更早期表现通常与更高的严重程度相关。1 其多系统累及可能需要多学科联合管理。
流行病学
由于 ARPKD 比 ADPKD 更少见,流行病学数据也相对较少。据报道,其发生率为每 26,500 例活产中 1 例,但可能在每 10,000 至 50,000 例活产中 1 例的范围内变化。1,17 然而,在孤立或近亲婚配的人群中可能更为常见。
发病机制
ARPKD 以经典的常染色体隐性方式遗传,未受累的杂合子父母生育受累子女的风险为25%。其病因为位于6号染色体上的多囊肾与肝疾病1(PKHD1)基因发生突变,该基因编码纤毛囊蛋白(亦称多导管蛋白),该蛋白定位于肾小管系统(集合管和亨利袢粗升支)、肝内胆管上皮细胞以及细胞顶端的原发性纤毛中。5,18,19
典型患者可在新生儿期就诊,既往有产前少羊水、肾脏增大,以及”Potter”序列伴肺发育不全,约30%的受累新生儿发生围产期死亡。20,21 增大的肾脏在超声上呈高回声,伴集合管梭形扩张,且可在不同年龄进展为ESRD。对其他器官系统的影响可表现为胆管扩张、先天性肝纤维化、门静脉高压以及神经认知功能障碍。22
评估与诊断
胎儿和新生儿超声可见由于大量微囊导致的双侧、显著增大且弥漫性高回声的肾脏。1 存在大囊肿(> 10 mm)可能提示其他纤毛病,如 MCDK 或 ADPKD;在严重羊水过少的情况下,MRI 可帮助进一步评估异常。13 准确诊断取决于提示性的影像学发现、具有隐性遗传方式的家族史、肝活检显示门周病变(在 ARPKD 中存在而在 ADPKD 中罕见),以及缺乏与其他囊性肾病相关的肾外表现。1
治疗选择、预后及并发症
与ADPKD类似,ARPKD目前无治愈方法,因此许多治疗方案旨在对与高血压、充血性心力衰竭以及肾衰竭和肝衰竭相关的症状进行管理或姑息处理。对于因肾脏增大产生占位效应而出现呼吸或营养受损的重症患者,可能需要采取积极治疗,例如单侧或双侧肾切除术。可实施减压治疗以处理门静脉高压。常考虑血液透析和肾移植。1
孤立性肾囊肿
孤立性和多发性肾囊肿是成人及老年人肾脏最常见的病变,但在儿童和婴儿中则罕见。4 它们可在产前即被发现,尽管产前病变常在妊娠期间自行消退。23
流行病学
产前超声显示肾囊肿的患病率为0.09%。23 出生至18岁期间单纯性囊肿的发生率平均为0.22%,范围为0.1%至0.45%。24 在成人及随年龄增长时发生率升高。
发病机制
大小差异很大,但多数小于 2 cm,且多位于肾皮质,可改变肾轮廓;也可位于深部皮质或看似位于肾髓质,但与肾盂无连续性。1 它们通常不影响肾功能,但当囊肿压迫邻近结构或阻塞肾集合系统时,偶尔会引起疼痛。5
评估与诊断
该病变通常为偶然发现,若符合以下标准,可通过超声安全地作出诊断:1) 内部无回声的囊肿,2) 薄且界限清楚的壁,边缘分明,3) 超声透声良好,囊后声增强,4) 形态呈球形或卵圆形。1 如不满足这些标准,或当超声对更复杂的囊肿等情况评估不理想时,CT扫描可提供更多解剖学细节。或者,可考虑MRI或穿刺抽吸。25
简单囊肿与肾盂不相通,然而有些囊肿可能看起来非常接近。对于是否存在相通的担忧,可通过静脉注射对比剂并包含延迟期的横断面成像进行评估,例如CT尿路造影或MR尿路造影。此外,囊肿穿刺抽吸及实验室检测可帮助识别是否与肾盂相通。若不相通,血尿素氮和肌酐将与患者的数值相当。5,26,27 在有阳性家族史的情况下,单发囊肿也可能引发对ADPKD的担忧。28 从历史上看,Bosniak囊肿分级在儿童中的验证并不充分,然而一项最新研究认为,改良的Bosniak分级系统可提供合理的风险分层,并显示mBosniak评分为1或2的病变往往为良性,而评分为3或4者有90%证实包含中间性或恶性病理。29
治疗方案及其疗效
有症状的囊肿可以行引流,然而若不使用硬化剂,复发的可能性较高。5 另一种处理选择是手术干预,进行囊肿去顶术,该术式适合采用微创的腹腔镜途径,尤其适用于相对表浅和/或外生性的囊肿。
随访可包括超声检查,以监测提示恶性的征象。稳定的囊肿若在两年内未见增大,可能无需进一步随访。29,30,31,32,33 在病变被证实为非恶性后,可能无需干预。
肾盏憩室
肾盏憩室(CD)是肾盏向肾实质突出的罕见囊状膨出。它们分为两型:1型更常见,毗邻上极或下极肾盏;2型较大,与肾盂相通,且更常有症状。34
流行病学
据报道,在接受静脉尿路造影(IV urogram)检查的儿童中,有 0.2% 至 0.6% 被发现存在 CD。13
发病机制
CD 的发病机制在很大程度上尚不清楚,且不被认为是一种遗传性疾病。然而,理论也包括基因突变可诱发影响肾脏的综合征,或输尿管芽回退失败,抑或梗阻性原因所致。13 囊肿内衬移行上皮,并与集合系统相通,典型地通过肾盏或变窄或狭窄的盏颈,更少见的是通过肾盂相通。35
评估与诊断
患者可能无症状,或表现为血尿、疼痛、尿路感染或肾结石;儿童最常见的表现是发热性尿路感染。35 无症状的憩室通常在影像学检查中被发现。尽管超声可初步识别肾盏憩室,但若要区分单纯性囊肿与肾盏憩室,最准确的影像学检查是使用静脉对比剂并包含延迟期的横断面影像学检查。35,36,37 图 1 和 图 2 显示了MR尿路造影上与CD一致的发现。近期一项将超声与横断面影像学检查进行比较的研究发现,针对性肾脏US的敏感性为40%、特异性为100%,而横断面影像学检查(具体为静态液体MR尿路造影)的敏感性为100%、特异性为91.6%。37 疼痛、感染、脓肿形成、泌尿源性败血症以及有症状的肾结石均可能成为手术治疗的适应证。
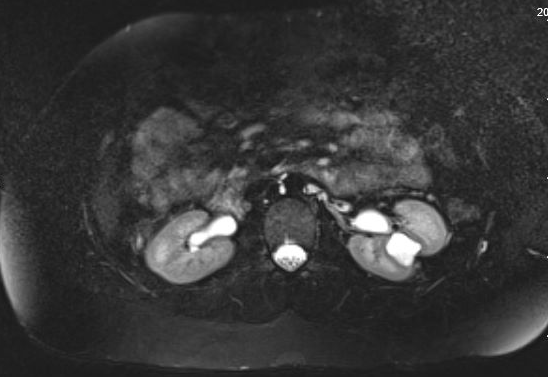
图 1 MR尿路造影显示左肾中极区的肾盏憩室被造影剂充盈,并见上覆肾实质变薄。
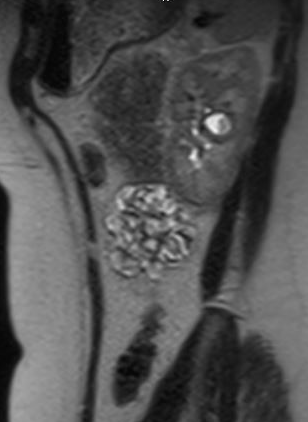
图 2 同一患者的MR尿路造影矢状位图像,显示造影剂充盈肾盏憩室。
治疗方案、其疗效及并发症
无症状患者不需要治疗,但可从超声监测中获益。1 对于具有手术处理指征者,如上所述,可能受益于腹腔镜下憩室袋形造口术并对上皮内衬进行电凝。38,39,40 其他可能的处理方式还包括输尿管镜术或微创经皮肾镜取石术(在存在结石的情况下),41 输尿管镜下腔体消融,39 或经皮消融与电凝。42 尽管治疗选择多样,针对CD的外科干预可能具有挑战性。为尽量降低并发症发生率,治疗通常从创伤更小的方案开始,如内镜或经皮途径。症状复发率可能较高,42 此时可将干预升级为腹腔镜/机器人手术,甚至开放手术,直至并包括部分肾切除或全肾切除。因此,在考虑对CD进行手术干预时,需要对患者进行充分的沟通与告知。
多囊性发育不良肾
多囊性发育不良肾(MCDK)是一种先天性畸形,常在儿童期或通过产前超声检查被诊断,其特征为单侧多发、互不相通的囊肿以及异常的后肾分化(可见软骨、未分化间充质和未成熟集合小管),常发生在尿路梗阻的背景下。4,13,43 双侧疾病通常因少羊水或无羊水导致的肺发育不全而与生命不相容。44
流行病学
MCDK是最常见的异常之一,估计发生率为每1,000至4,000例出生中有1例。1,45 鉴于真性肾缺如的发生率很低,成年期单肾的较高发生率很可能与MCDK随时间的消退有关。5
发病机制
多囊性发育不良发生在肾单位形成之前,其来源于后肾分化异常、肾母细胞异常,或肾发育期间出现的高级别梗阻。1 囊肿由低立方上皮内衬,周围为梭形细胞包绕,内容物可为蛋白样或血性液体。1 可见原始和发育不良成分,以及未成熟软骨或未成熟肾小球。MCDK 分为漏斗-肾盂型,其特点为肾盂与输尿管闭锁,和较少见的肾积水型,仅累及输尿管上段闭锁。46 对侧肾可出现代偿性肥大。
评估与诊断
目前,MCDK最常在产前超声检查中被发现,而其次常见的就诊情形是对新生儿可触及肿块进行评估。47
MCDK 的诊断必须与由其他情况(如肾盂输尿管连接部[UPJ]梗阻)导致的肾积水加以鉴别。值得注意的是,在 MCDK 中,囊肿为非交通性,且较大的囊肿多位于外侧。肾积水时,肾实质围绕中央的囊性结构,且典型的肾形外观得以保留。48 图3 和 图4 有助于展示在超声上将 MCDK 与肾积水鉴别的挑战。图5 展示了大小不一且彼此不相交通的囊肿,这是 MCDK 的典型表现。为进一步明确,可进行放射性核素显像;在肾积水时可见摄取,而在 MCDK 情况下通常仅有极少或无摄取。49
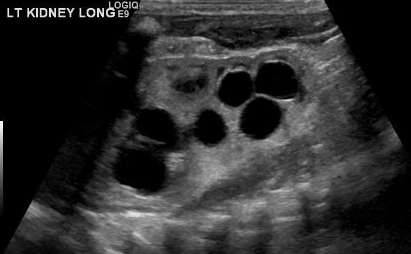
图 3 肾脏超声显示可能为MCDK的多个肾囊肿,或为肾盂肾盏扩张/肾积水(HDN)。
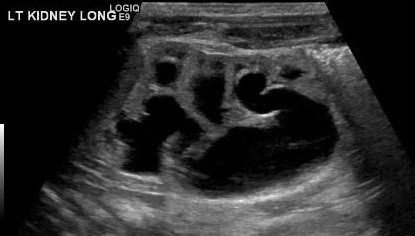
图 4 同一患者的另一张肾脏超声图像,显示肾盂与肾盏之间的连续性,与肾盂肾盏扩张/HDN一致。
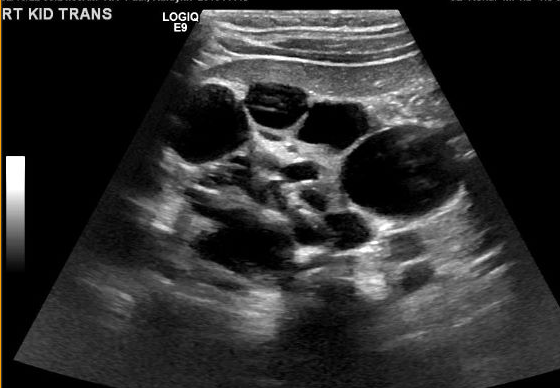
图 5 肾脏超声显示多个互不相通、大小不一的囊肿,与多囊性发育不良肾(MCDK)一致。
治疗方案、疗效及并发症
多囊性发育不良肾(MCDK)的自然病程表现为随时间逐渐退化,并可能出现包括高血压和恶性肿瘤在内的并发症。5 然而,后者的风险可能较低。50 从历史上看,受累肾脏曾因担忧罕见的恶性转化而被手术切除,但现已转向非手术管理,并通过超声监测双肾。手术指征包括提示发展为肾母细胞瘤(Wilms瘤)的可疑改变、占位效应、疼痛、高血压以及家长意愿。5 对于患有MCDK且对侧肾正常的患者,应在不损伤对侧肾的前提下,向其说明预计长期肾功能可维持正常,以给予安慰。还应就预防性维护措施的重要性进行宣教,例如在参与运动时使用防护装备,以及采取健康的生活方式(饮食和运动),以减轻其他肾脏疾病的致病因素(如高血压和糖尿病)。为评估慢性肾病发生的风险,随访还可包括对侧肾影像学检查、行排尿性膀胱尿道造影以筛查膀胱输尿管返流(VUR)、血压监测以及尿液分析以检测蛋白尿。51
髓质海绵肾
髓质海绵肾(MSK)以远端集合管扩张、囊肿和憩室为特征,且均局限于肾髓质锥体。1
流行病学
MSK主要见于青年期,但也可见于儿童。52 由于许多MSK患者无症状,其发病率尚不明确。在有钙性结石病史的人群中更为常见,并与先天性综合征有关,如贝克威思-威德曼综合征、埃勒斯-当洛综合征、无牙症和卡罗利病。1
发病机制
MSK 普遍被认为是非遗传性的,然而,也有病例似乎以常染色体显性方式遗传,这支持一种假说,即 MSK 破坏了输尿管芽与后肾胚基之间的界面。52
扩张的乳头内集合管和髓质囊肿内含钙化或脱落物质,使肾脏呈海绵样外观,并且在70%的病例中为双侧。1 囊肿与集合小管相连续,并由集合管上皮衬里。53
评估与诊断
诊断可依据尿路造影的表现:可见肾脏增大,偶有钙化,尤以肾乳头部位为著;肾乳头小管增宽并被造影剂充盈;以及乳头部对比剂渗染伴残余肾髓质充影。54 如诊断仍不明确,可通过肝脏评估在 MSK 与 ADPKD 之间加以区分。1
治疗方案、其结局和并发症
治疗策略主要取决于对结石或感染的处理。随后应进行结石预防措施,包括增加液体摄入、低钠饮食、噻嗪类利尿剂以及枸橼酸钾。52 与MSK相关的并发症与肾结石发生率增加及其相关的疾病负担有关。
肾单位萎缩症
肾单位消耗症(NPHP)是儿童慢性肾脏病的常见病因之一,呈隐性遗传,因其影像学和组织学表现相似,常与髓质囊性肾病(MCKD)并提;然而,MCKD为常染色体显性遗传,起病较晚(成人期),且无肾外受累。5
流行病学
NPHP 根据存在的基因缺陷分为婴儿期(1岁)、少年期(13岁)或青少年期(19岁)。婴儿期 NPHP 在 1–3 岁时即进展至终末期肾病(ESRD),并与 NPHP2 突变相关(亦称为 NPHP 2 型)。少年期 NPHP 是最常见的表现,在 10–13 岁时进展至终末期肾病,且最常与 NPHP1 突变相关(亦称为 NPHP 1 型)。最后,青少年期 NPHP 与 NPHP3 突变相关(亦称为 NPHP 3 型)。5,55
发病机制
尽管可见散发病例,NPHP 通常以常染色体隐性方式遗传,其中位于第2号染色体上的 NPHP1 基因的纯合致病性突变是最常见的原因。56 已知有许多基因与 NPHP 相关(参见(表 2)5
表 2 NPHP 的遗传异质性。5
| 类型 | 染色体定位 | 突变基因 |
|---|---|---|
| NPH 1型 | 2q13 | NPHP1 |
| NPH 2型 | 9q22 | NPHP2 |
| NPH 3型 | 3q22 | NPHP3 |
| NPH 4型 | 1p36 | NPHP4 |
| NPH 5型 | 3q21 | NPHP5 |
| PH 6型 | 12q21 | NPHP6 |
| NPH 7型 | 16p | NPHP7 |
| NPH 8型 | 16q | NPHP8 |
| NPH 9型 | 17q11 | NPHP9 |
| NPH 10型 | 1q44 | SDCCAG8 |
| PH 11型 | 8q22 | MKS3 |
| AHI1 | 6q23 | AHI1 |
NPHP 肾脏的大体观察可因类型不同而异,但通常体积较小,表面呈颗粒样。组织学上,可见间质纤维化和单核细胞浸润,肾小管改变多样,包括萎缩、基底膜增厚及层状化。57 NPHP 的囊肿起源于远曲小管和集合管。57
评估、诊断与并发症
尿浓缩能力受损可导致多尿和多饮(脱水、夜尿),并导致肾性钠丢失;出现CKD的症状,如乏力、食欲减退、生长迟缓;以及肾性骨营养不良的症状。可能出现与肾功能衰竭程度不相称的贫血。除2型NPHP外,高血压(HTN)较少见。5,58 肾外受累包括肝脏和门静脉纤维化并伴肝肿大,以及许多其他表现:Senior-Løken综合征(NPHP和视网膜营养不良)、骨骼异常、智力发育迟缓、小脑性共济失调、内脏反位、心脏畸形,或其他较少见的综合征。5
超声检查是常用的检测手段,但可能无法检出所有病例;薄层计算机断层扫描 (CT) 可检出此类病例。58 当影像学检查结果不明确时,可通过基因检测以及肾脏组织学检查进行诊断。建议进行肝脏和眼部筛查,以评估肝纤维化和Senior-Løken综合征。5
治疗方案及其结局
针对 NPHP,目前可用的有效治疗方法很少。当前的管理包括针对 CKD 的支持性和预防性管理。在肾功能衰竭时,更倾向于进行移植,因为供肾中不会发生肾小管损伤。13
多房性囊肿
多房性囊性肾瘤(cystic nephroma)是一种发生于成人和儿童的良性肿瘤性病变,与Wilms瘤处于同一疾病谱。
流行病学
大多数病例发生在4岁之前或30岁之后,且在儿科人群中以男性为主。59
发病机制
这些病变体积庞大,互不相通,由纤维组织良好包裹,可能含有胚胎组织,并可能压迫正常肾实质。5
评估、诊断与治疗方案
儿童多房性囊肿最常见的表现为可触及的腹部包块。60 评估通常采用超声或CT扫描,然而,由于仅凭影像有时难以区分多房性囊肿与囊性肾母细胞瘤(Wilms瘤),最终诊断可能需要在行肾切除或部分肾切除后通过组织病理学检查作出。60
获得性囊性肾病
获得性囊性肾病(ACKD)指在终末期肾病(ESRD)和氮质血症背景下出现的双侧肾脏的囊性改变,而非遗传性肾囊性疾病。1
流行病学
在终末期肾病(ESRD)患者中,10%可发现囊肿;在开始透析后3年和5年,这一比例分别增加至44%和60%。1
发病机制
病因被认为与毒素介导、生长因子积聚,或由于纤维化、草酸盐结晶、血管闭塞或缺血导致的肾小管阻塞有关。1 双侧肾脏均受累,且大小通常较正常为小;组织学显示为与远端肾小管上皮相似的扁平至立方上皮。4
评估、诊断、治疗和并发症
大多数囊肿患者无症状,但由于囊内自发性出血,可能出现疼痛和/或血尿。超声检查常用于诊断与监测,而CT或MRI可能更易识别囊肿,然而在终末期肾病(ESRD)患者中,因对比剂具有肾毒性作用,静脉注射对比剂的影像学检查应谨慎进行。1 主要并发症为出血和肿瘤性转化。4 建议进行随访性超声检查,在怀疑肿瘤时可行肾切除。5 因此,治疗以保守为主,采取症状管理并调整筛查计划或透析方案。
与囊性肾病相关的综合征
肾囊肿可见于多种综合征,其中最常见的可能包括常染色体显性遗传的结节性硬化症和冯-希佩尔-林道病。其他呈常染色体隐性遗传的包括梅克尔综合征、Jeune 窒息性胸发育不良,以及泽尔韦格脑-肝-肾综合征。1
结节性硬化症
结节性硬化症的诊断依据为:符合两条主要标准,或一条主要标准加两条次要标准(见表3)。61 多发性肾囊肿被列为一项次要标准。61,62 在结节性硬化症中,肾脏受累是导致发病率和死亡率的重要原因。63
表 3 结节性硬化症的主要与次要诊断标准。61
| 主要标准 | 次要标准 |
|---|---|
| 低色素斑(≥3,直径至少5 mm) | “五彩纸屑样”皮损 |
| 血管纤维瘤(≥3)或头面部纤维性斑块 | 牙釉质凹陷(>3) |
| 甲周纤维瘤(≥2) | 口腔内纤维瘤(≥2) |
| 鲨皮样斑块 | 视网膜无色素斑 |
| 多发性视网膜错构瘤 | 多发性肾囊肿 |
| 皮质发育异常 | 非肾性错构瘤 |
| 室管膜下结节 | |
| 室管膜下巨细胞星形细胞瘤 | |
| 心脏横纹肌瘤 | |
| 淋巴管肌瘤病(LAM) | |
| 血管平滑肌脂肪瘤(≥2) |
冯·希佩尔-林道综合征
冯·希佩尔-林道综合征同样具有多种表现,其中肾细胞癌和肾囊肿较为常见,但在儿童中较少见。64
梅克尔综合征
这可能表现为多种表现形式,包括囊肿。囊性发育不良必然存在。5,65
结论
液体充填的肾囊肿见于一大类异质性的肾囊性疾病。尽管它们共同表现为肾囊肿,这组疾病可通过多种因素加以区分:遗传方式、起病年龄、伴随的肾内或肾外异常、临床病程、组织学差异、影像学表现等。由于症候学表现的重叠,准确识别具体的疾病过程可能具有挑战性。影像学技术,尤其是超声检查,长期以来一直是识别其中许多疾病的主要方式。基因检测选择正变得越来越普遍且更易获得。
持续的研究推动基础科学和临床进展,有助于发现新的疾病、病理生理机制、诊断手段和治疗方案。
要点
- 区分单纯性囊肿与肾盏憩室可能较为困难,但尤为重要,因为肾盏憩室更可能引起症状并需要干预。诊断可借助静脉对比增强的横断面影像及延迟期成像来加以促进。
- 同样重要的是区分多囊性发育不良肾(MCDK)与重度肾积水,例如在肾盂输尿管连接部(UPJ)梗阻时。MCDK 的处理主要以观察为主,而对肾积水或 UPJ 梗阻更有可能需要进行干预。诊断可借助放射性核素显像加以促进。
- 某些囊性肾病(如常染色体显性多囊肾病[ADPKD]和常染色体隐性多囊肾病[ARPKD])的管理应与肾脏科协作进行,以实现最佳的内科肾脏照护。
- 产前影像对囊肿的特征评估常常有助于区分可能的疾病(见下方的图 6)。13

图 6 通过产前影像对囊肿进行鉴别诊断.13
建议阅读
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
参考文献
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- Glassberg KI, Stephens FD, Lebowitz RL. Renal dysgenesis and cystic disease of the kidney: a report of the Committee on Terminology, Nomenclature and Classification, Section on Urology, American Academy of Pediatrics. J Urol Oct 1987; 138 (4 Pt 2): 1085–1092. DOI: 10.1016/s0022-5347(17)43510-5.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364 (16): 1533–1543. DOI: 10.1056/NEJMra1010172.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Cole BR, Conley SB, Stapleton FB. Polycystic kidney disease in the first year of life. J Pediatr Nov 1987; 111 (5): 693–699. DOI: 10.1016/s0022-3476(87)80244-5.
- Gabow PA. Polycystic kidney disease: clues to pathogenesis. Kidney Int Dec 1991; 40 (6): 989–996. DOI: 10.1038/ki.1991.306.
- Proesmans W, Damme B, Casaer P, Marchal G. Autosomal dominant polycystic kidney disease in the neonatal period: association with a cerebral arteriovenous malformation. Pediatrics Dec 1982; 70 (6): 971–975. DOI: 10.1542/peds.70.6.971.
- Ong AC, Wheatley DN. Polycystic kidney disease–the ciliary connection. Lancet Mar 2003; 361 (9359): 774–776. DOI: 10.1016/S0140-6736(03)12662-1.
- Rossetti S, Consugar MB, Chapman AB. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol Jul 2007; 18 (7): 2143–2160. DOI: 10.1681/ASN.2006121387.
- Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol Oct 2004; 15 (10): 2528–2536. DOI: 10.1097/01.ASN.0000141055.57643.E0.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet Apr 1994; 343 (8901): 824–827. DOI: 10.1016/s0140-6736(94)92026-5.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Grantham JJ. Polycystic kidney disease: hereditary and acquired. Adv Intern Med 1993; 38: 409–420.
- Schrier R, McFann K, Johnson A. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol Jul 2002; 13 (7): 1733–1739. DOI: 10.1097/01.asn.0000018407.60002.b9.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med Oct 1990; 323 (16): 1091–1096. DOI: 10.1056/NEJM199010183231602.
- Alzarka B, Morizono H, Bollman JW, Kim D, Guay-Woodford LM. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Front Pediatr 2017; 5 (80). DOI: 10.3389/fped.2017.00080.
- Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers 2018; 4 (1). DOI: 10.1038/s41572-018-0047-y.
- Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatr Nephrol Jan 1989; 3 (1): 43–49. DOI: 10.1007/BF00859625.
- Ward CJ, Hogan MC, Rossetti S. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet Mar 2002; 30 (3): 259–269. DOI: 10.1038/ng833.
- Onuchic LF, Furu L, Nagasawa Y. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet May 2002; 70 (5): 1305–1317. DOI: 10.1086/340448.
- Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol Jun 1997; 11 (3): 302–306. DOI: 10.1007/s004670050281.
- Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics Sep 2014; 134 (3). DOI: 10.1542/peds.2013-3646.
- Blazer S, Zimmer EZ, Blumenfeld Z, Zelikovic I, Bronshtein M. Natural history of fetal simple renal cysts detected in early pregnancy. J Urol 1999; 162 (3 Pt 1): 812–814. DOI: 10.1097/00005392-199909010-00066.
- McHugh K, Stringer DA, Hebert D, Babiak CA. Simple renal cysts in children: diagnosis and follow-up with US. Radiology Feb 1991; 178 (2): 383–385. DOI: 10.1148/radiology.178.2.1987597.
- Bosniak MA. The current radiological approach to renal cysts. Radiology Jan 1986; 158 (1): 1–10. DOI: 10.1148/radiology.158.1.3510019.
- Lee J, Darcy M. Renal cysts and urinomas. Semin Intervent Radiol. Dec 2011; 28 (4): 380–391. DOI: 10.1055/s-0031-1296080.
- Steinhardt GF, Slovis TL, Perlmutter AD. Simple renal cysts in infants. Radiology May 1985; 155 (2): 349–350. DOI: 10.1148/radiology.155.2.3885305.
- Gabow PA, Kimberling WJ, Strain JD, Manco-Johnson ML, Johnson AM. Utility of ultrasonography in the diagnosis of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol Jan 1997; 8 (1): 105–110. DOI: 10.1681/ASN.V81105.
- Peard L, Gargollo P, Grant C. Validation of the modified Bosniak classification system to risk stratify pediatric cystic renal masses: An international, multi-site study from the pediatric urologic oncology working group of the societies for pediatric urology. J Pediatr Urol 2022; 18 (2). DOI: 10.1016/j.jpurol.2021.12.001.
- Bayram MT, Alaygut D, Soylu A, Serdaroğlu E, Cakmakçı H, Kavukçu S. Clinical and radiological course of simple renal cysts in children. Urology Feb 2014; 83 (2): 433–437. DOI: 10.1016/j.urology.2013.08.055.
- Karmazyn B, Tawadros A, Delaney LR. Ultrasound classification of solitary renal cysts in children. J Pediatr Urol Jun 2015; 11 (3). DOI: 10.1016/j.jpurol.2015.03.001.
- O’Kelly F, McAlpine K, Abdeen N, Keays MA, Leonard MP, Guerra LA. The Prevalence, Clinicodemographics, and Outcomes of Incidental and Symptomatic Renal Cysts in a Pediatric Cohort Undergoing Ultrasonography. J Urol 2019; 202 (2): 394–399. DOI: 10.1097/JU.0000000000000264.
- Rediger C, Guerra LA, Keays MA. Renal cyst evolution in childhood: a contemporary observational study. J Pediatr Urol Apr 2019; 15 (2). DOI: 10.1016/j.jpurol.2019.01.006.
- Wulfsohn MA. Pyelocaliceal diverticula. J Urol Jan 1980; 123 (1): 1–8. DOI: 10.1016/s0022-5347(17)55748-1.
- Estrada CR, Datta S, Schneck FX, Bauer SB, Peters CA, Retik AB. Caliceal diverticula in children: natural history and management. J Urol Mar 2009; 181 (3): 1311. DOI: 10.1016/j.juro.2008.10.043.
- Waingankar N, Hayek S, Smith AD, Okeke Z. Calyceal diverticula: a comprehensive review. Rev Urol 2014; 16 (1): 29–43.
- Sahin H, Sarioglu FC, Alaygut D, Akdogan AI, Pekcevik Y. Differentiation of simple renal parenchymal cyst and calyceal diverticulum. Pediatr Int May 2020; 62 (5): 615–623. DOI: 10.1111/ped.14127.
- Casale P, Grady RW, Feng WC, Joyner BD, Mitchell ME. The pediatric caliceal diverticulum: diagnosis and laparoscopic management. J Endourol Sep 2004; 18 (7): 668–671. DOI: 10.1089/end.2004.18.668.
- Long CJ, Weiss DA, Kolon TF, Srinivasan AK, Shukla AR. Pediatric calyceal diverticulum treatment: An experience with endoscopic and laparoscopic approaches. J Pediatr Urol Aug 2015; 11 (4). DOI: 10.1016/j.jpurol.2015.04.013.
- Sripathi V, Mitra A, Padankatti RL, Ganesan T. Robotic treatment of a type 2 calyceal diverticulum in a child: is suture closure and marsupialisation enough for a good outcome? J Robot Surg Dec 2018; 12 (4): 727–730. DOI: 10.1007/s11701-017-0758-1.
- Ding X, Xu ST, Huang YH. Management of symptomatic caliceal diverticular calculi: Minimally invasive percutaneous nephrolithotomy versus flexible ureterorenoscopy. Chronic Dis Transl Med Dec 2016; 2 (4): 250–256. DOI: 10.1016/j.cdtm.2016.11.016.
- Monga M, Smith R, Ferral H, Thomas R. Percutaneous ablation of caliceal diverticulum: long-term followup. J Urol Jan 2000; 163 (1): 28–32. DOI: 10.1016/s0022-5347(05)67965-7.
- Gilbert-Barness E, Potter EL. Respiratory system. 2nd ed., DOI: 10.1001/jama.1997.03540320078045.
- D’Alton M, Romero R, Grannum P, DePalma L, Jeanty P, Hobbins JC. Antenatal diagnosis of renal anomalies with ultrasound. IV Bilateral Multicystic Kidney Disease Am J Obstet Gynecol Mar 1986; 154 (3): 532–537. DOI: 10.1016/0002-9378(86)90597-1.
- Kalyoussef E, Hwang J, Prasad V, Barone J. Segmental multicystic dysplastic kidney in children. Urology Nov 2006; 68 (5). DOI: 10.1016/j.urology.2006.06.024.
- Felson B, Cussen LJ. The hydronephrotic type of unilateral congenital multicystic disease of the kidney. Semin Roentgenol Apr 1975; 10 (2): 113–123. DOI: 10.1016/0037-198x(75)90035-8.
- Welch TR, Wacksman J. The changing approach to multicystic dysplastic kidney in children. J Pediatr Jun 2005; 146 (6): 723–725. DOI: 10.1016/j.jpeds.2005.02.027.
- Sanders RC, Hartman DS. The sonographic distinction between neonatal multicystic kidney and hydronephrosis. Radiology Jun 1984; 151 (3): 621–625. DOI: 10.1148/radiology.151.3.6718720.
- Roach PJ, Paltiel HJ, Perez-Atayde A, Tello RJ, Davis RT, Treves ST. Renal dysplasia in infants: appearance on 99mTc DMSA scintigraphy. Pediatr Radiol 1995; 25 (6): 472–475. DOI: 10.1007/BF02019071.
- Narchi H. Risk of Wilms’ tumour with multicystic kidney disease: a systematic review. Arch Dis Child Feb 2005; 90 (2): 147–149. DOI: 10.1136/adc.2004.051243.
- Mansoor O, Chandar J, Rodriguez MM. Long-term risk of chronic kidney disease in unilateral multicystic dysplastic kidney. Pediatr Nephrol Apr 2011; 26 (4): 597–603. DOI: 10.1007/s00467-010-1746-0.
- Gambaro G, Danza FM, Fabris A. Medullary sponge kidney. Curr Opin Nephrol Hypertens. Jul 2013; 22 (4): 421–426. DOI: 10.1097/MNH.0b013e3283622b86.
- Bernstein J. The classification of renal cysts. Nephron 1973; 11 (2): 91–100. DOI: 10.1159/000180222.
- Gedroyc WM, Saxton HM. More medullary sponge variants. Clin Radiol Jul 1988; 39 (4): 423–425. DOI: 10.1016/s0009-9260(88)80292-7.
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol Jan 2009; 20 (1): 23–35. DOI: 10.1681/ASN.2008050456.
- Hildebrandt F, Otto E, Rensing C. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet Oct 1997; 17 (2): 149–153. DOI: 10.1038/ng1097-149.
- Sherman FE, Studnicki FM, Fetterman G. Renal lesions of familial juvenile nephronophthisis examined by microdissection. Am J Clin Pathol Apr 1971; 55 (4): 391–400. DOI: 10.1093/ajcp/55.4.391.
- Elzouki AY, al-Suhaibani H, Mirza K, al-Sowailem AM. Thin-section computed tomography scans detect medullary cysts in patients believed to have juvenile nephronophthisis. Am J Kidney Dis Feb 1996; 27 (2): 216–219. DOI: 10.1016/s0272-6386(96)90543-0.
- Eble JN, Bonsib SM. Extensively cystic renal neoplasms: cystic nephroma, cystic partially differentiated nephroblastoma, multilocular cystic renal cell carcinoma, and cystic hamartoma of renal pelvis. Semin Diagn Pathol Feb 1998; 15 (1): 2–20.
- Castillo OA, Boyle ET, Kramer SA. Multilocular cysts of kidney. A study of 29 patients and review of literature. Urology Feb 1991; 37 (2): 156–162. DOI: 10.1016/0090-4295(91)80214-r.
- Northrup H, Krueger DA, ITSCC G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol Oct 2013; 49 (4): 243–254. DOI: 10.1016/j.pediatrneurol.2013.08.001.
- Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol Dec 1998; 13 (12): 624–628. DOI: 10.1177/088307389801301206.
- Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc Aug 1991; 66 (8): 792–796. DOI: 10.1016/s0025-6196(12)61196-3.
- Maddock IR, Moran A, Maher ER. A genetic register for von Hippel-Lindau disease. J Med Genet Feb 1996; 33 (2): 120–127. DOI: 10.1136/jmg.33.2.120.
- Salonen R. The Meckel syndrome: clinicopathological findings in 67 patients. Am J Med Genet Aug 1984; 18 (4): 671–689. DOI: 10.1002/ajmg.1320180414.
最近更新时间: 2025-09-22 08:00