
40: DSD—État actuel des connaissances, bilan et traitement
Ce chapitre prendra environ 25 minutes de lecture.
Introduction et terminologie
Les troubles/différences du développement sexuel (DSD, également désignés sous le terme d’intersexuation) sont des affections congénitales dans lesquelles le sexe chromosomique, gonadique ou phénotypique diffère de ce qui est considéré comme typiquement masculin ou féminin. Une nouvelle classification large des DSD a été introduite en 2006 via le “Consensus Statement on Management of Intersex Disorders.”1 Cette classification plus récente intègre un large éventail d’affections, notamment l’hyperplasie congénitale des surrénales (CAH), les DSD ovotesticulaires, le syndrome d’insensibilité aux androgènes (AIS). La définition consensuelle de 2006 est plus vaste que ce que l’on désignait auparavant sous le nom d’affections intersexuelles, incluant également des anomalies anatomiques telles que l’exstrophie cloacale et vésicale et l’agénésie vaginale, ainsi que des anomalies chromosomiques qui n’entraînent pas de génitalité atypique (p. ex., syndrome de Klinefelter [47, XXY]). Tableau 1 présente un aperçu de la nomenclature antérieure et des mises à jour terminologiques recommandées proposées dans l’énoncé de consensus de 2006. Étant donné la grande variété d’affections relevant du ‘parapluie’ DSD, une approche de prise en charge individualisée et multidisciplinaire est primordiale.
Tableau 1 Nomenclature DSD révisée. Source : Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
| Précédent | Proposé |
|---|---|
| Intersexe | DSD |
| Pseudo-hermaphrodite mâle | 46,XY DSD |
| Pseudo-hermaphrodite féminin | 46,XX DSD |
| Vrai hermaphrodite | DSD ovotesticulaire |
| Dysgénésie gonadique mixte | Dysgénésie gonadique mixte (inchangé) |
| Homme XX ou inversion sexuelle XX | DSD testiculaire 46,XX |
| Inversion sexuelle XY | Dysgénésie gonadique complète 46,XY |
Depuis son introduction, la nouvelle terminologie DSD a été adoptée de façon quasi universelle par la communauté médicale. Parmi les personnes concernées par les affections DSD, il existe une variabilité des préférences terminologiques, et il n’y a pas de consensus quant aux affections qui devraient exactement être considérées comme un DSD.2,3,4 En particulier, certains membres de la communauté CAH s’identifient comme présentant un trouble endocrinien, et non une affection DSD/intersexe.5 On ne sait pas non plus quelles personnes atteintes d’un hypospadias proximal devraient être considérées comme ayant « un DSD ».6 Les cliniciens qui prennent en charge des affections DSD doivent être conscients de l’évolution de la nomenclature et des controverses, et utiliser les termes que leurs patients préfèrent lors des consultations médicales individuelles. Aux fins de ce chapitre, la terminologie actuelle, médicalement acceptée, sera utilisée.
Embryologie
Développement typique des organes génitaux internes/externes
Tous les fœtus en développement commencent de la même manière. Les structures anatomiques nécessaires au développement masculin ou féminin, ainsi qu’à leurs variations, sont présentes dès les premières semaines de gestation (Tableau 2), y compris la crête gonadique, les canaux de Wolff (mésonéphriques) et de Müller (paramésonéphriques), le cloaque et le sinus urogénital qui en dérive, le tubercule génital et les bourrelets labio-scrotaux. Figure 1 montre les gènes responsables du développement de la gonade indifférenciée en un testicule ou un ovaire.
Tableau 2 Précurseurs embryonnaires et structures typiques masculines/féminines.
| Structure embryologique | Structure typique féminine | Structure typique masculine |
|---|---|---|
| Crête gonadique | Ovaire | Testicule |
| Canal de Wolff (mésonéphrique) | Paroophoron, époophoron, kyste du canal de Gartner | Canal déférent, épididyme, vésicules séminales |
| Canal de Müller (paramésonéphrique) | Trompes de Fallope, utérus, vagin proximal | Appendice testiculaire, utricule prostatique |
| Cloaque et sinus urogénital subséquent | Vessie, vagin distal, urètre | Vessie, prostate, urètre |
| Tubercule génital | Clitoris | Pénis |
| Bourrelets labioscrotaux | Complexe labial | Scrotum |
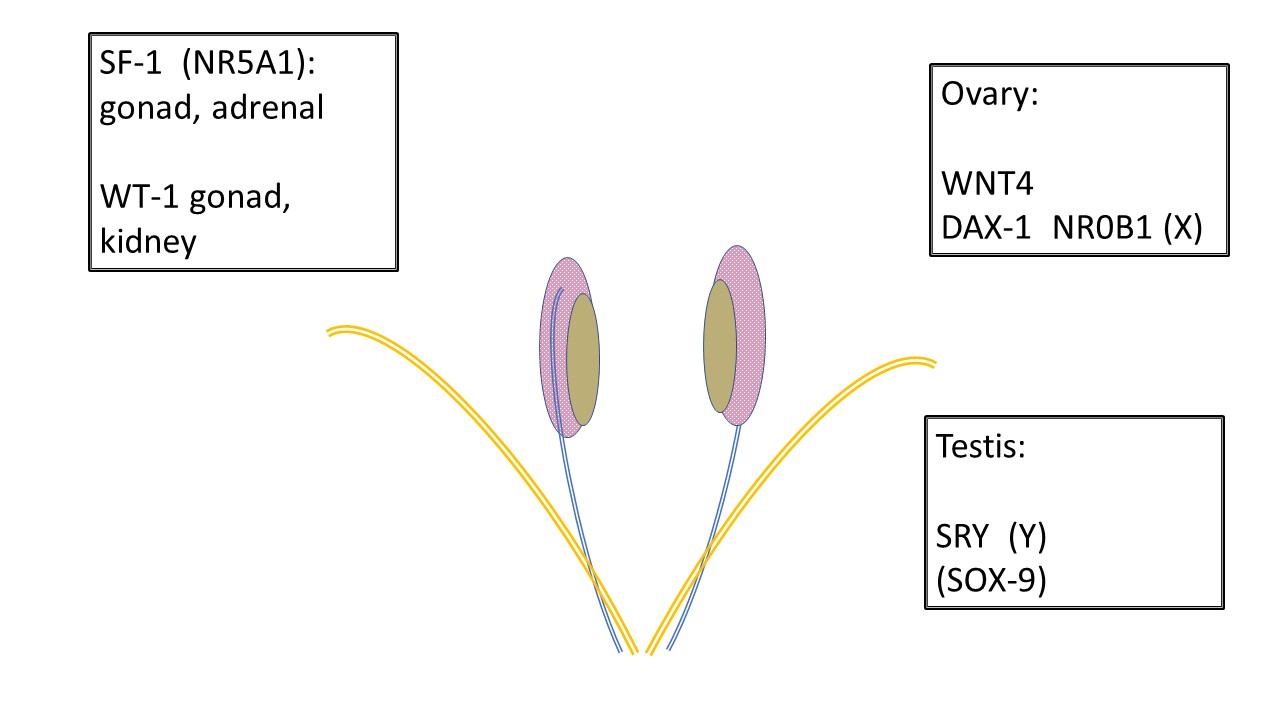
Figure 1 Développement des gonades indifférenciées. Toutes les gonades débutent de manière identique. Les cellules germinales primordiales migrent vers la crête gonadique avant 6 semaines et l’infrastructure soutenant le développement gonadique est en outre influencée par divers gènes. SF-1 et WT-1 influencent le développement gonadique et la communication endocrine subséquente vers les canaux de Wolff (mésonéphriques-bleu) et de Müller (paramésonéphriques-orange). Noter la relation entre le mésonéphros (rose), le canal mésonéphrique et la gonade indifférenciée (beige). WNT4 est le gène de la détermination ovarienne. DAX-1 est considéré comme le “gène anti-testiculaire”. La duplication de DAX-1 entraîne une inversion sexuelle XY. SRY sur le chromosome Y est le gène de la détermination testiculaire. SOX-9 soutient le développement des cellules de Sertoli mais possède également une homologie lui permettant de participer au développement testiculaire en l’absence de SRY.
Le développement masculin typique (Figure 2) est initié très tôt au cours de la gestation par le chromosome Y, en particulier SRY, qui détermine le développement testiculaire. WT-1, SF-1 interviennent dans le développement du testicule et de l’ovaire, et SOX-9 est impliqué dans la différenciation des cellules de Sertoli. Les cellules de Leydig du testicule commencent ensuite la production de testostérone à environ 8 semaines de gestation et les cellules de Sertoli produisent l’hormone anti-müllérienne (AMH), inhibant la maturation des canaux de Müller en utérus, trompes de Fallope et partie supérieure du vagin. La testostérone dicte la croissance précoce du tubercule génital et le développement du canal de Wolff en canal déférent, épididyme et vésicule séminale. Le développement, ou son absence, dans les systèmes wolffien et müllérien se produit par une action paracrine plutôt que systémique, ce qui explique un développement asymétrique des structures canalaires lorsque la composition gonadique est asymétrique. La conversion de la testostérone en dihydrotestostérone (DHT), plus puissante, par la 5α-réductase-2 a lieu dans divers tissus et est responsable de la maturation ultérieure du pénis, de la tubulisation du sinus urogénital et de la plaque urétrale, ainsi que de la fermeture du scrotum à 14 semaines de gestation. La croissance ultérieure de la structure phallique survient en raison de la diminution des androgènes fœtaux et de la croissance somatique.
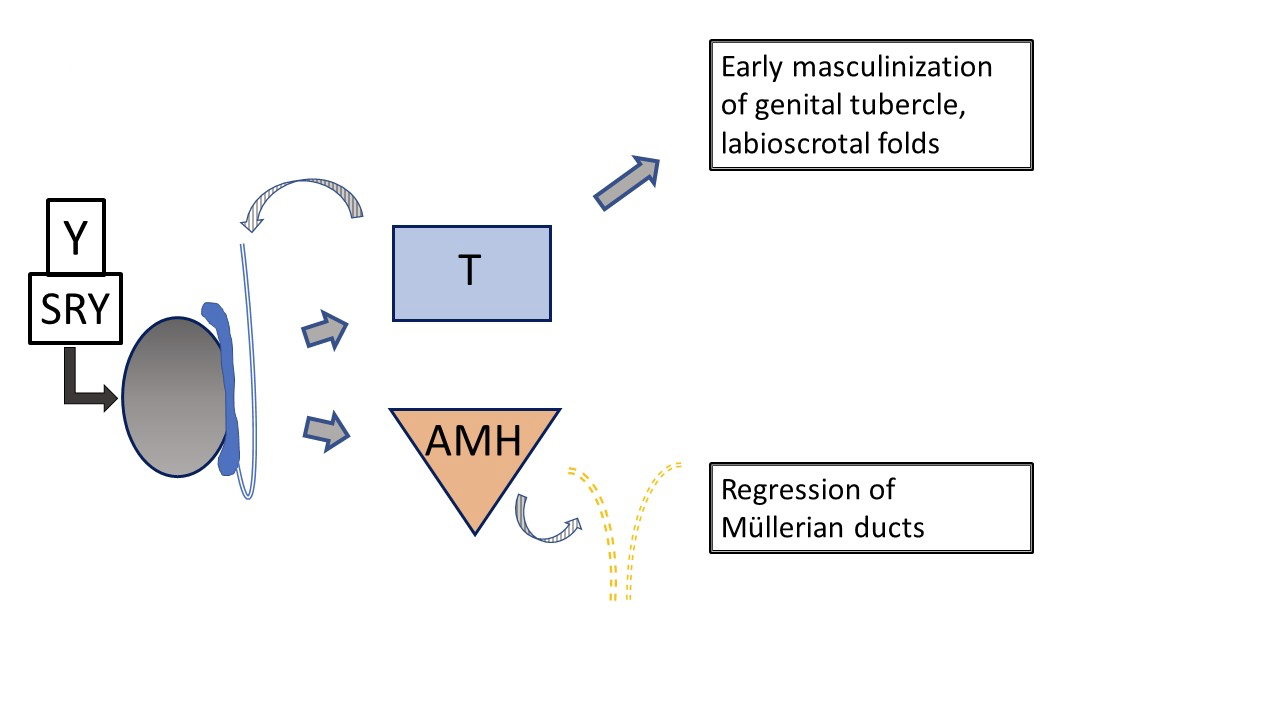
Figure 2 Développement précoce typique du tractus génital masculin externe et interne, tel qu’influencé par le testicule fœtal. La production de testostérone et d’AMH par le testicule fœtal entraîne une masculinisation précoce du tubercule génital et la régression des canaux de Müller. Le développement ultérieur des organes génitaux externes et de l’urètre est influencé par la conversion de la testostérone en dihydrotestostérone. (non représenté ici, voir Figure 4).
Le développement féminin typique (Figure 3) est souvent considéré comme se produisant par défaut puisqu’il n’est pas sous la régulation des hormones produites par les testicules. La détermination ovarienne ne joue pas un rôle majeur dans le développement embryologique des organes génitaux. De faibles niveaux d’androgènes et d’AMH permettent le développement féminin typique du tubercule génital en clitoris, des plis labioscrotais en complexe labial, et du sinus urogénital distal en urètre et en vagin. Les canaux de Müller appariés migrent et fusionnent pour former l’utérus. Ensemble, ils stimulent le bulbe sinovaginal au niveau du sinus urogénital pour initier le développement de la portion inférieure du vagin.
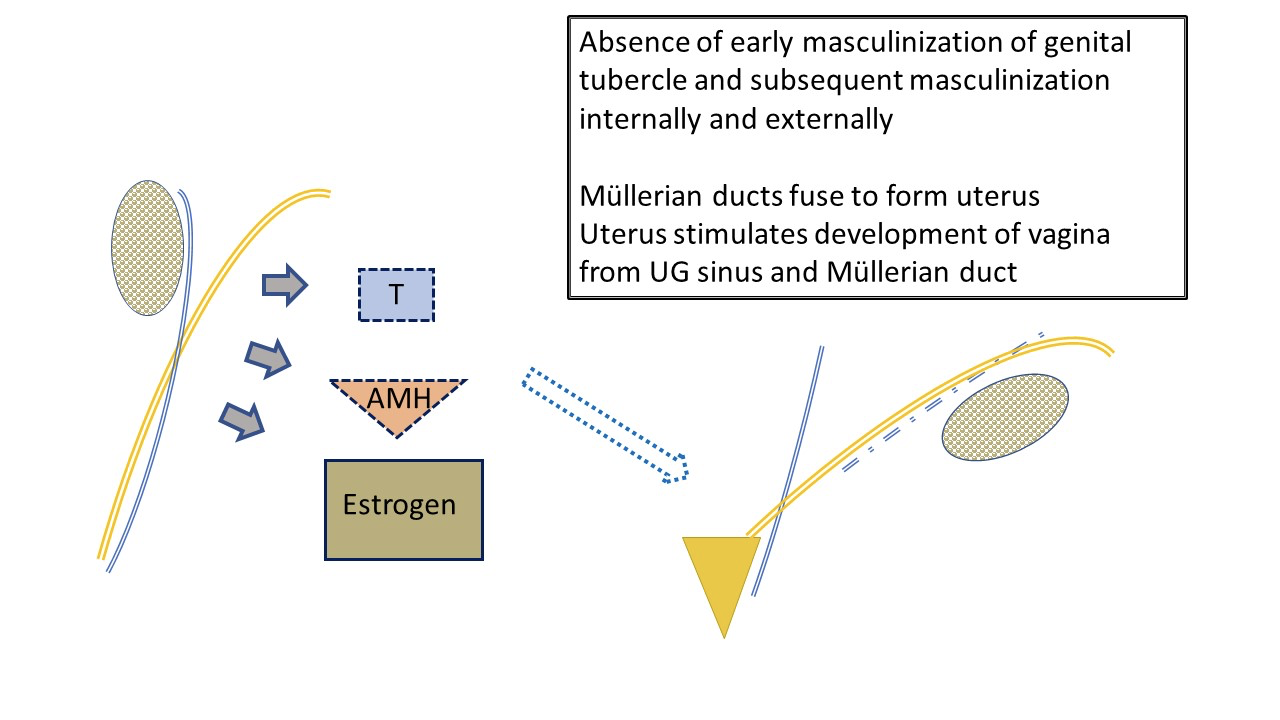
Figure 3 Développement précoce typique de l’appareil génital féminin externe et interne, sous l’influence de l’ovaire fœtal. De faibles niveaux d’AMH et d’androgènes produits par l’ovaire entraînent le développement d’une anatomie féminine typique : utérus et trompes de Fallope, vagin supérieur. La production fœtale d’œstrogènes a moins d’impact sur le développement de la filière féminine que l’environnement caractérisé par des niveaux minimaux d’androgènes et d’AMH. Les lèvres seront influencées par les œstrogènes maternels. Noter également la relation finale “eau sous le pont” entre l’uretère (issu du canal mésonéphrotique) et la trompe de Fallope. Noter les reliquats du canal mésonéphrotique associés à la trompe de Fallope.
Différentes voies pouvant être à l’origine d’un DSD
Les différences du développement peuvent survenir de manière atypique :
- Détermination génétique des gonades (p. ex., dysgénésie gonadique mixte, DSD ovotesticulaire)
- Production hormonale (p. ex., déficit en 5α-réductase, déficit en 3-β-hydroxystéroïde déshydrogénase, CAH par déficit en 21-hydroxylase, ou mutations du récepteur de l’hormone lutéinisante [LH])
- Action hormonale (p. ex., AIS complet ou partiel)
- Variations du développement des tissus précurseurs/séparation du cloaque (p. ex., syndrome de Mayer-Rokitansky-Küster-Hauser [MRKH], exstrophie cloacale, anomalies cloaquales)
Même avec un caryotype 46,XX ou 46,XY, il n’y a aucune certitude que le développement suivra la voie féminine ou masculine typique. Chez les 46,XY uniquement, de nombreuses différences surviennent en raison de mutations du chromosome Y qui altèrent la détermination ou la fonction testiculaire, de mutations somatiques entraînant une dysgénésie du testicule en développement, de variations de la biosynthèse ou de la conversion de la testostérone, et de mutations liées à l’X qui affectent la fonction du récepteur des androgènes. Le phénotype externe, le développement interne, le potentiel de puberté spontanée ou de fertilité, ainsi que l’identité de genre sur le spectre, peuvent tous être affectés. En examinant les caractéristiques des divers troubles du développement sexuel (DSD) décrits ci-dessous, il est important de noter que la production et l’action hormonales s’inscrivent sur un continuum. Ainsi, les déficits de fonction des récepteurs ou de production d’hormones et d’enzymes de conversion ne sont pas nécessairement complets. Le phénotype peut varier largement, même entre des individus ayant le même diagnostic.
Physiopathologie
Les variations du développement des organes génitaux internes et externes sont déterminées par des mutations des chromosomes sexuels et des mutations somatiques, la fonction gonadique qui en résulte et la réponse tissulaire. Lorsque la fonction d’un récepteur ou la présence d’une enzyme pertinente est atypique en raison d’une mutation génétique connue ou encore non identifiée, l’effet sur les organes cibles peut être complet (absence de fonction évidente) ou partiel. Le phénotype varie en conséquence. À titre d’exemple, divers phénotypes d’insensibilité aux androgènes (CAIS complet, PAIS partiel et MAIS léger: spectre du phénotype génital allant du féminin typique—ambigu—masculin typique) résultent de nombreuses mutations distinctes du gène du récepteur aux androgènes sur le chromosome X (affection récessive liée à l’X). Tableau 3 présente un système de classification des DSD, divisé selon le caryotype.
Tableau 3 Un exemple de classification des DSD
| TDS des chromosomes sexuels | TDS 46,XY | TDS 46,XX |
|---|---|---|
| 45,X (syndrome de Turner et variantes) | Troubles du développement gonadique (testiculaire) : (1) dysgénésie gonadique complète (syndrome de Swyer) ; (2) dysgénésie gonadique partielle ; (3) régression gonadique ; et (4) TDS ovotesticulaire |
Troubles du développement gonadique (ovarien) : (1) TDS ovotesticulaire ; (2) TDS testiculaire (p. ex., SRY+, duplication de SOX9) ; et (3) dysgénésie gonadique |
| 47,XXY (syndrome de Klinefelter et variantes) | Troubles de la synthèse ou de l’action des androgènes : (1) défaut de biosynthèse des androgènes (p. ex., déficit en 17-hydroxystéroïde déshydrogénase, déficit en 5RD2, mutations de StAR) ; (2) défaut d’action des androgènes (p. ex., CAIS, PAIS) ; (3) défauts du récepteur de l’hormone lutéinisante (p. ex., hypoplasie, aplasie des cellules de Leydig) ; et (4)troubles de l’hormone anti-Müllérienne et du récepteur de l’hormone anti-Müllérienne (syndrome des canaux de Müller persistants) |
Excès d’androgènes : (1) fœtal (p. ex., déficit en 21-hydroxylase, déficit en 11-hydroxylase) ; (2) fœtoplacentaire (déficit en aromatase, POR [oxydoréductase P450]) ; et (3) maternel (lutéome, exogène, etc) |
| 45,X/46,XY (MGD, TDS ovotesticulaire) | Autres (p. ex., exstrophie cloacale, atrésie vaginale, MURCS [anomalies müllériennes, rénales, des somites cervicothoraciques], autres syndromes) | |
| 46,XX/46,XY (chimérique, TDS ovotesticulaire) |
46,XX TDS
La forme la plus courante de TDS 46,XX est l’HCS, dans laquelle la fonction enzymatique surrénalienne est altérée. Figure 4 montre la cascade stéroïdogénique surrénalienne typique, qui commence par le cholestérol. Les produits finaux sont l’aldostérone, le cortisol et les stéroïdes sexuels. La branche des stéroïdes sexuels de la cascade se termine par l’androstènedione, qui est convertie en une petite quantité de testostérone. La production de testostérone dans les cellules de Leydig du testicule commence également avec le cholestérol. Dans l’HCS, la diminution de la fonction enzymatique entraîne une baisse des minéralocorticoïdes (aldostérone) et des glucocorticoïdes (cortisol), ainsi qu’un détournement vers la production de stéroïdes sexuels. Il en résulte une virilisation d’un fœtus au caryotype 46,XX. Bien qu’il soit plus simple de retenir une absence complète de fonction enzymatique et de produits stéroïdiens surrénaliens en aval, la fonction peut n’être que partiellement altérée et la rétroaction au niveau de l’axe hypothalamo-hypophysaire s’en trouver affectée en conséquence.
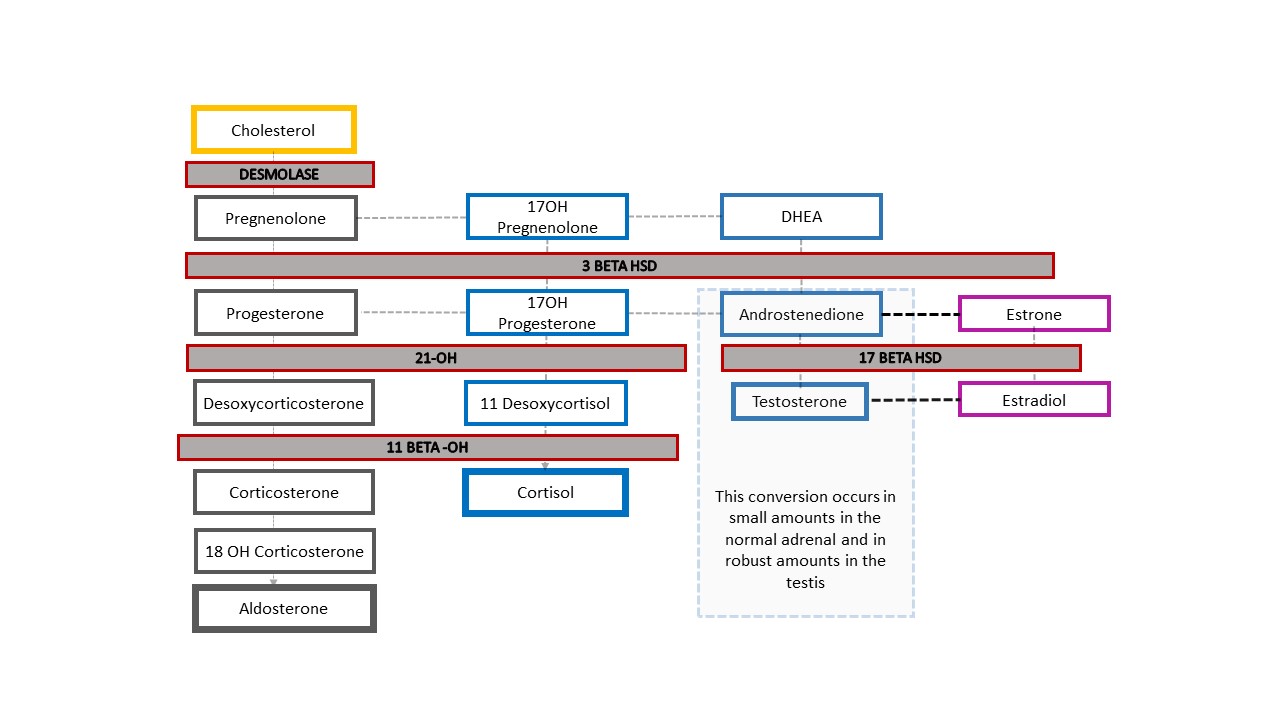
Figure 4 Cascade stéroïdienne et spectre des diagnostics d’HCS. Tout degré de défaut de production d’une enzyme se traduit par une diminution du produit en aval et un détournement de la cascade vers les stéroïdes sexuels. L’axe hypothalamo-hypophyso-surrénalien est stimulé pour compenser. L’augmentation de l’ACTH entraîne une hyperplasie du cortex surrénalien et une augmentation supplémentaire de la production de précurseurs.
L’anatomie typique d’un patient atteint d’HCS 46,XX comprend des degrés variables d’hypertrophie clitoridienne et la formation d’un sinus urogénital. Les gonades sont des ovaires et ne sont pas palpables. Il existe plusieurs mutations qui conduisent à différents milieux stéroïdiens surrénaliens caractéristiques décrits dans les manuels et à des phénotypes physiques et physiologiques. La forme la plus fréquente est le déficit en 21-hydroxylase. Les nourrissons atteints d’HCS 46,XX peuvent présenter une perte de sel mettant en jeu le pronostic vital vers l’âge d’une semaine de vie, de sorte que ce diagnostic doit être envisagé chez tous les nourrissons présentant des organes génitaux ambigus. Une élévation de la 17‑OH‑progestérone est diagnostique. L’identité de genre féminine et un sexe d’éducation féminin sont les plus fréquents, bien que non universels.
46,XY TDS
Dans la catégorie des TDS 46,XY, il existe un éventail d’anomalies du développement gonadique et de la synthèse et de l’action des androgènes, comme détaillé dans Tableau 3. En plus du spectre des formes de SIA détaillées ci‑dessus, la dysgénésie gonadique complète ou partielle peut entraîner des variations anatomiques similaires : un nourrisson porteur de chromosomes 46,XY et présentant soit un phénotype féminin typique, soit un phénotype ambigu (p. ex., hypospadias proximal avec transposition pénoscrotale, et une ou les deux gonades non descendues). Les personnes présentant une SIA complète et celles présentant une dysgénésie gonadique complète sont généralement élevées comme filles et s’identifient comme femmes ; celles présentant des formes partielles de l’une ou l’autre affection montrent une variabilité plus grande du sexe d’éducation et de l’identité de genre.
Une autre forme de DSD 46,XY, le déficit en 5α‑réductase, entraîne une conversion réduite de la testostérone en dihydrotestostérone (DHT), ce qui se traduit par un phénotype génital variable. Figure 5 et Figure 6 comparent la voie de développement testiculaire typique à celle qui survient chez les patients atteints d’un déficit en 5α‑réductase.
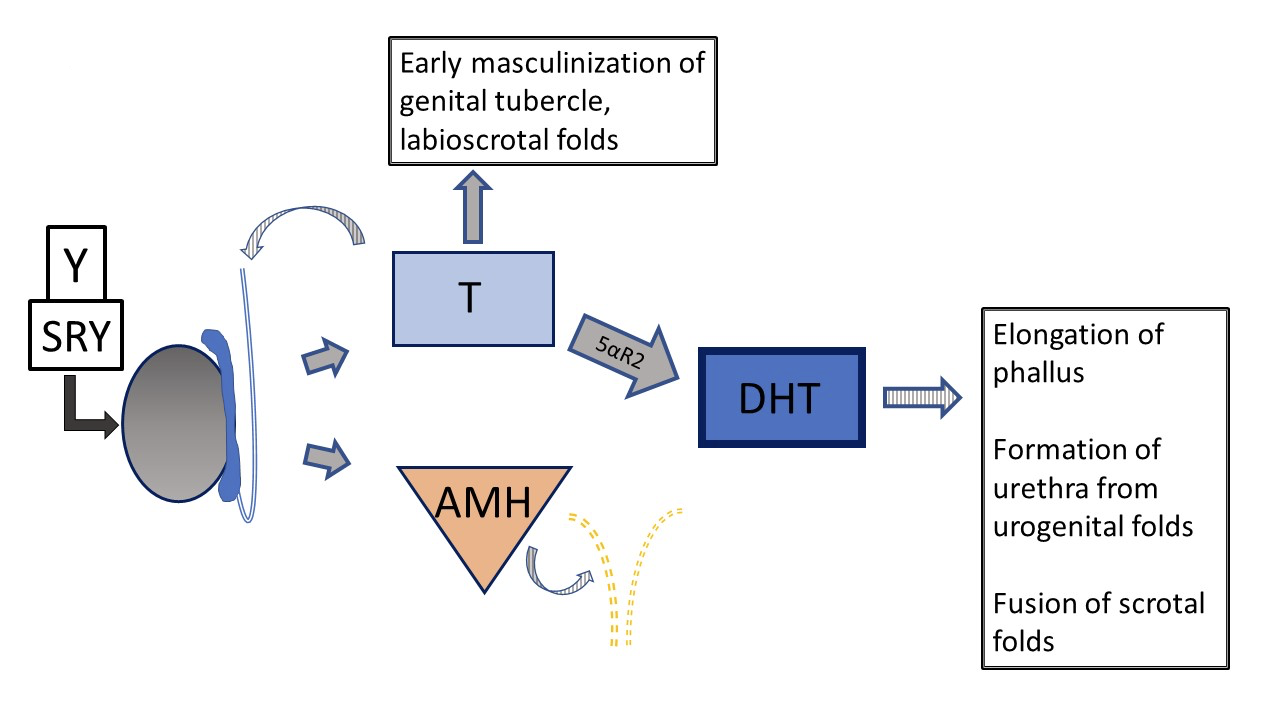
Figure 5 Développement génital prénatal 46XY typique Le développement peut s’écarter du “typique” à plusieurs étapes différentes. Les flèches hachurées indiquent une fonction de type paracrine. Les lignes en pointillés indiquent la régression des canaux de Müller au début du développement.
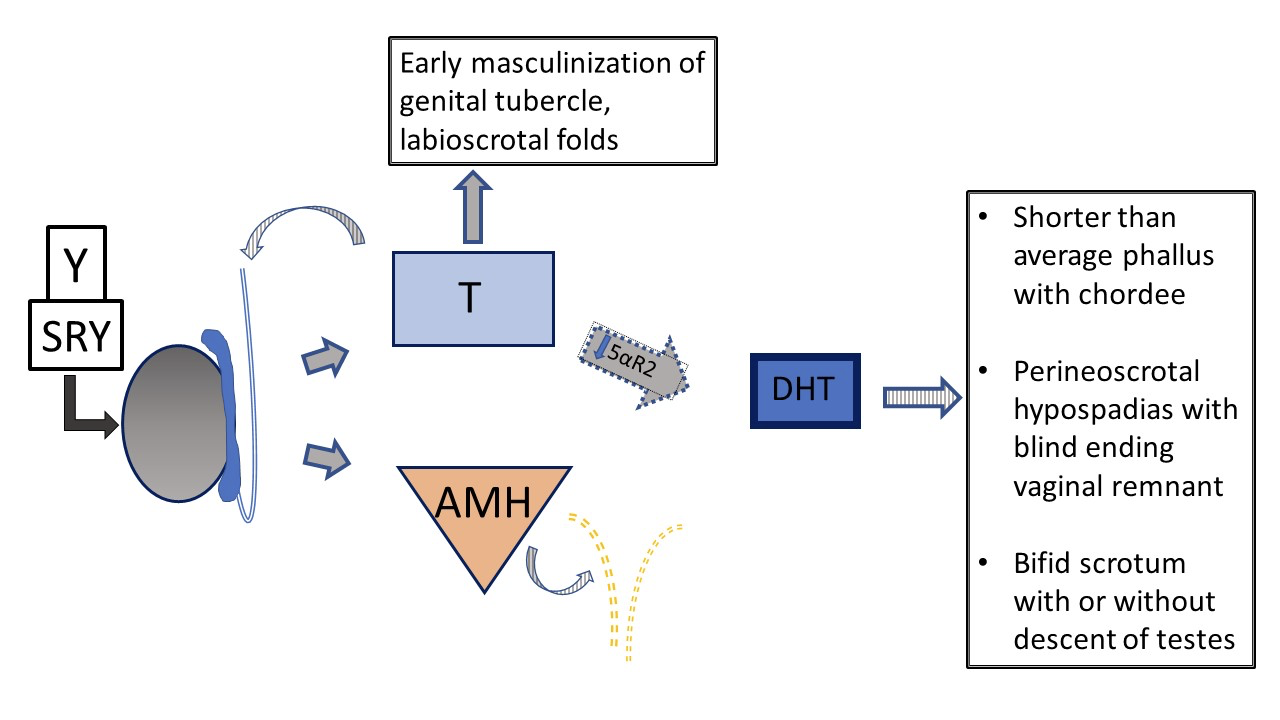
Figure 6 Développement génital prénatal 46XY chez des patients avec vs déficit en 5-α-réductase. Les testicules sont différenciés et présentent une production normale de testostérone et d’AMH. Il n’y a pas d’utérus. En raison d’une altération de la fonction de la 5-α-réductase, le rapport T /DHT est nettement augmenté. Dans un contexte de DHT abaissée, on observe une variabilité du phénotype allant d’un féminin typique à un masculin typique, jusqu’à une anatomie génitale ambiguë. En cas de déficit sévère, le développement du phallus et du sinus urogénital est arrêté à un stade précoce de la masculinisation, avec un orifice urogénital unique dans la région périnéale et l’absence de fusion des plis labioscrotaux.
Figure 7 montre la cascade surrénalienne résultante dans l’exemple rare d’un patient 46,XY atteint de CAH se présentant cliniquement avec des organes génitaux ambigus : déficit en 3-β-hydroxystéroïde déshydrogénase [HSD]. Le blocage enzymatique survient avant la conversion de la déhydroépiandrostérone (DHEA) en androstènedione dans la cascade stéroïdienne surrénalienne, de sorte que les nourrissons présentent une diminution des minéralocorticoïdes et des glucocorticoïdes ainsi qu’une différence du développement génital. Comme les nourrissons 46,XX atteints de CAH par déficit en 21-hydroxylase ou en 11β-hydroxylase, les nourrissons présentant un déficit en 3-β-HSD peuvent présenter une perte de sel engageant le pronostic vital. Le diagnostic est établi par l’élévation de la 17-OH prégnénolone et de la DHEA.
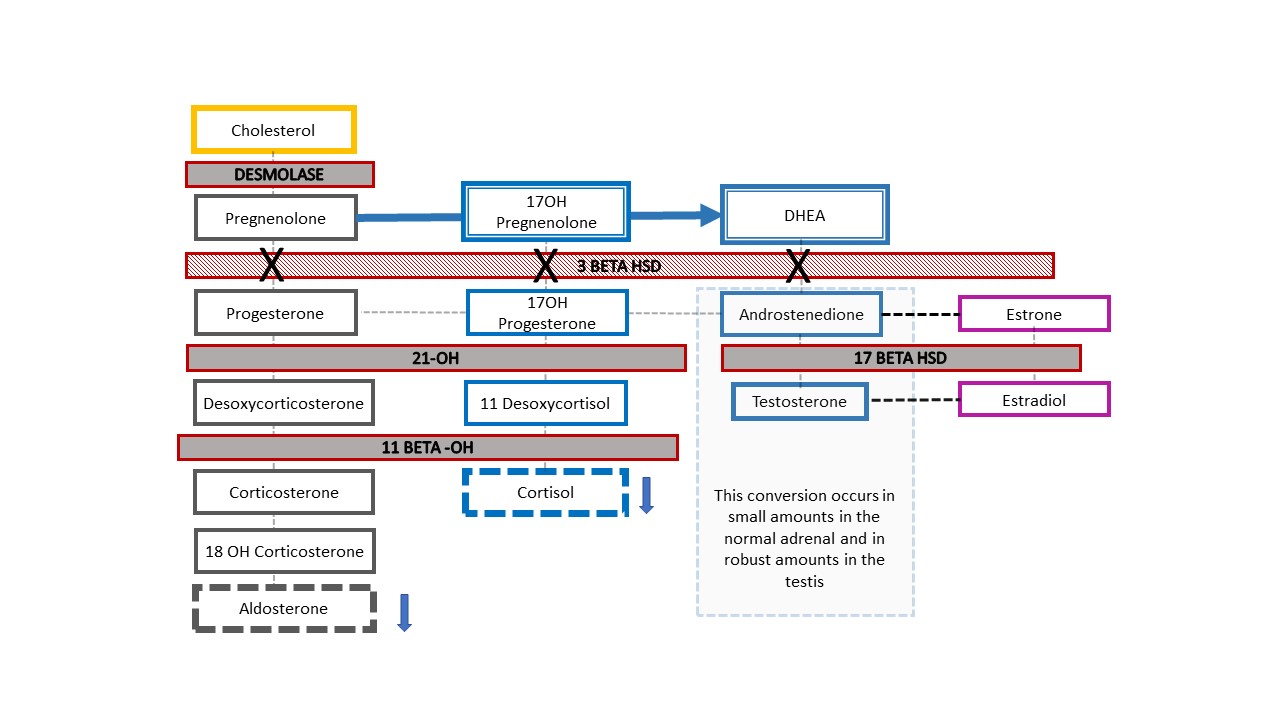
Figure 7 Voie de stéroïdogenèse en cas de déficit en 3-β-hydroxystéroïde déshydrogénase. Ce défaut relativement proximal dans la cascade du cholestérol surrénalien entraîne un déficit sévère en glucocorticoïdes et en minéralocorticoïdes. Il est unique, en ce que les nouveau-nés 46,XY atteints d’HCS présentent un aspect génital sous-masculinisé, tandis que les 46,XX ont un aspect génital féminin typique (ou quasi typique). Lorsqu’un nouveau-né avec chromosome Y détecté en prénatal naît avec un aspect ambigu des organes génitaux, c’est le type d’HCS à exclure en premier lieu. Des taux élevés de 17-OH-prégnénolone et de DHEA aident à poser le diagnostic.
DSD liés aux chromosomes sexuels
Parmi les diagnostics de DSD des chromosomes sexuels listés dans Tableau 3, les affections avec un caryotype mosaïque 45,X/46,XY (ou similaire) méritent une mention particulière. Les patients présentant ce caryotype peuvent avoir des organes génitaux externes d’apparence féminine typique (syndrome de Turner avec matériel chromosomique Y [TS+Y]), des organes génitaux ambigus (dysgénésie gonadique mixte [MGD]) ou des organes génitaux externes d’apparence masculine typique. Les patients avec TS+Y ont un risque accru de tumeurs gonadiques, ce qui diffère des patients atteints d’un syndrome de Turner 45,X. Le phénotype et le sexe d’éducation/identité de genre sont variables dans la MGD, bien que l’on observe fréquemment une asymétrie gonadique et labio-scrotale, comme détaillé dans Figure 8. Tout patient avec un caryotype 45,X/46,XY peut présenter des manifestations du TS (p. ex., coarctation aortique, anomalies rénales), et doit donc subir les mêmes tests de dépistage systémique que les patients atteints de TS classique.7,8
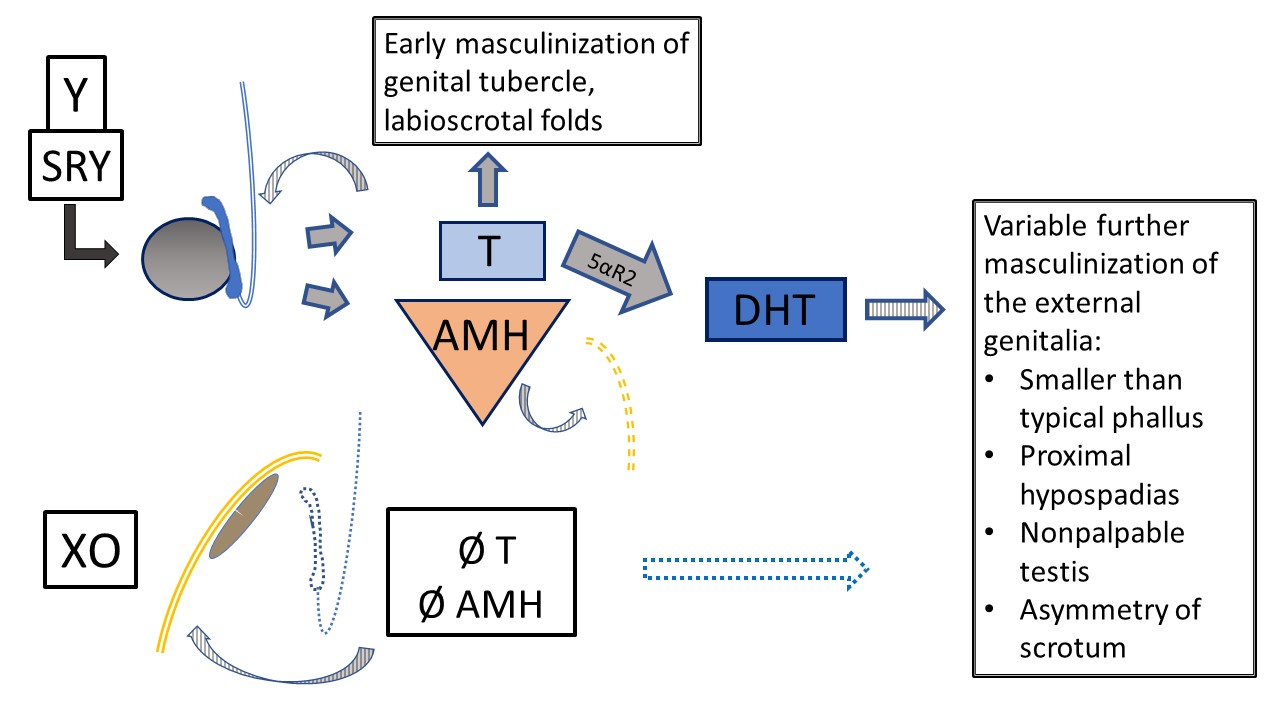
Figure 8 Dysgénésie gonadique mixte 45,X/46,XY. Plusieurs phénotypes surviennent avec ce caryotype en mosaïque. Ce schéma représente le développement interne et externe dans le contexte d’un testicule (déterminé par le chromosome Y) et d’une gonade en bandelette (développement par défaut, stroma de type ovarien). Le phénotype classique, lorsqu’un degré d’ambiguïté est observé, est une gonade non palpable, un testicule descendu, et un hypospadias proximal. Comme la T et l’AMH ont une fonction paracrine, la gonade en bandelette est associée à un hémi-utérus et à une trompe de Fallope sans structures wolffiennes. En raison du matériel Y, il existe un risque de gonadoblastome et de dysgerminome. Le testicule présente un développement dysgénétique variable, impactant la fonction précoce et future ainsi que le risque tumoral. Une virilisation primaire survient mais le développement ultérieur de l’urètre et du scrotum est variablement influencé par la fonction testiculaire.
TDS ovotesticulaire
Le DSD ovotesticulaire survient lorsqu’il existe à la fois du tissu ovarien et du tissu testiculaire chez le même individu (ancien nom – hermaphrodisme vrai). Généralement, le tissu ovarien est mieux développé, et le tissu testiculaire est dysgénétique. Le phénotype, le sexe d’éducation/l’identité de genre et le potentiel de fertilité sont tous variables. Fait important, les patients atteints de DSD ovotesticulaire peuvent présenter n’importe quel caryotype (bien que 46, XX soit le plus fréquent), de sorte que ce diagnostic doit être envisagé chez tout patient qui se présente avec des organes génitaux ambigus.
Évaluation multidisciplinaire
Au-delà de proposer une nomenclature moins péjorative, d’autres pierres angulaires de la déclaration de consensus de 2006 sont paradigme sont l’accompagnement par une équipe multidisciplinaire et le soutien psychosocial longitudinal des patients et des parents.1
L’équipe multidisciplinaire
L’équipe qui travaille avec une famille lors d’une consultation pour un nouveau-né hospitalisé est de préférence la même qui suivra les patients et les familles au long cours. L’équipe est responsable d’informer la famille sur la pathologie et sur toutes les options de prise en charge envisageables ; elle apporte un soutien et associe la famille à une prise de décision partagée lorsque des décisions sont nécessaires (p. ex., sexe d’éducation, divulgation, chirurgie). Idéalement, l’équipe comprend les membres suivants :
- Endocrinologie
- Santé mentale (Psychologue, Travailleur social)
- Urologie / Chirurgie pédiatrique / Gynécologie
- Conseiller en génétique / Généticien
- Soins infirmiers
- Représentant des patients / Éducateur des patients
- Néonatologue (consultation auprès de patients hospitalisés)
Chaque spécialiste apporte au sein de l’équipe pluridisciplinaire des perspectives différentes, une expertise clinique pertinente et des compétences interpersonnelles. Les chirurgiens constituent une partie importante de l’équipe, même si les indications chirurgicales précoces ou urgentes sont peu fréquentes. Tous prennent part aux discussions d’équipe et contribuent à l’expérience du patient et de sa famille. Idéalement, le respect mutuel et la volonté de prendre en compte toutes les perspectives améliorent le fonctionnement de l’équipe et lui permettent de fournir, au fil du temps, des soins d’un niveau encore plus élevé, en particulier à mesure que les standards de soins évoluent.
Chaque personne vivant avec une variation du développement sexuel (VDS) peut ne pas avoir accès à une équipe multidisciplinaire fonctionnelle. Certaines peuvent ne pas adhérer à la prise en charge par l’équipe ou trouver le suivi longitudinal contraignant. Chaque occasion précoce d’information sur le diagnostic et sur les préoccupations potentielles à long terme ou les besoins médicaux est donc très importante. Dans les situations où un suivi à long terme est faisable et acceptable, cela peut être réalisé dans le cadre d’une prise en charge plus régulière par une seule spécialité, selon ce qui convient au patient. Par exemple, certains patients atteints d’HCS au sein de l’institution des auteurs sont suivis trimestriellement par leur endocrinologue et participent à une visite complète en équipe multidisciplinaire une fois par an.
Travailler activement ensemble, apprendre les uns des autres et des patients/familles, facilite le fonctionnement de l’équipe et fait progresser la prise en charge. L’équipe s’appuie sur des expériences antérieures et assimile de nouvelles connaissances, en particulier dans les domaines de l’expérience patient et des analyses génétiques de nouvelle génération. L’équipe identifie également des lacunes dans les connaissances et dans le paradigme de la prise en charge qui peuvent être comblées grâce à des collaborations et à une recherche ciblée. Des conférences multidisciplinaires périodiques basées sur des cas ou des thématiques aident les membres de l’équipe et les apprenants à renforcer leur maîtrise des différentes pathologies et offrent l’occasion de comprendre les différences de philosophie.
Approche initiale du patient chez qui un trouble du développement sexuel (TDS) est suspecté
Les patients chez qui l’on suspecte une DSD se présentent classiquement dans la petite enfance avec des organes génitaux ambigus, ou à l’adolescence avec une aménorrhée primaire. Les affections DSD peuvent également attirer l’attention médicale à la suite de découvertes fortuites en imagerie ou peropératoires, d’une hernie inguinale chez une fille, ou si une fille présente une masculinisation au moment de la puberté. De plus en plus, les DSD sont suspectées en prénatal en raison d’une discordance entre les résultats du test d’ADN libre circulant et ceux de l’échographie, ou d’une inquiétude quant à une différence du développement génital observée à l’échographie.
Lorsqu’un nourrisson chez qui un TDS est suspecté naît, il est recommandé, chaque fois que possible, de consulter une équipe multidisciplinaire (décrite ci-dessus). N’oubliez pas de féliciter la famille pour la naissance de leur bébé (une étape souvent négligée, surtout lorsqu’il y a surprise ou manque de familiarité concernant les TDS). Utilisez des termes neutres du point de vue du genre, (“votre bébé”, pas “it”) pour faire référence au nourrisson s’il existe une question quant à l’assignation du sexe. Figure 9 présente des recommandations pour décrire l’examen génital du nouveau-né, y compris des termes neutres à utiliser. En plus d’une anamnèse prénatale détaillée, des antécédents familiaux et de l’examen physique, les recommandations pour l’évaluation initiale d’un patient chez qui un TDS est suspecté comprennent :
- Caryotype
- Bilan endocrinien (17-OH-progestérone, testostérone, hormone lutéinisante, hormone folliculostimulante, électrolytes)
- Échographie pelvienne pour évaluer la position et le type des gonades, ainsi que la présence de structures müllériennes
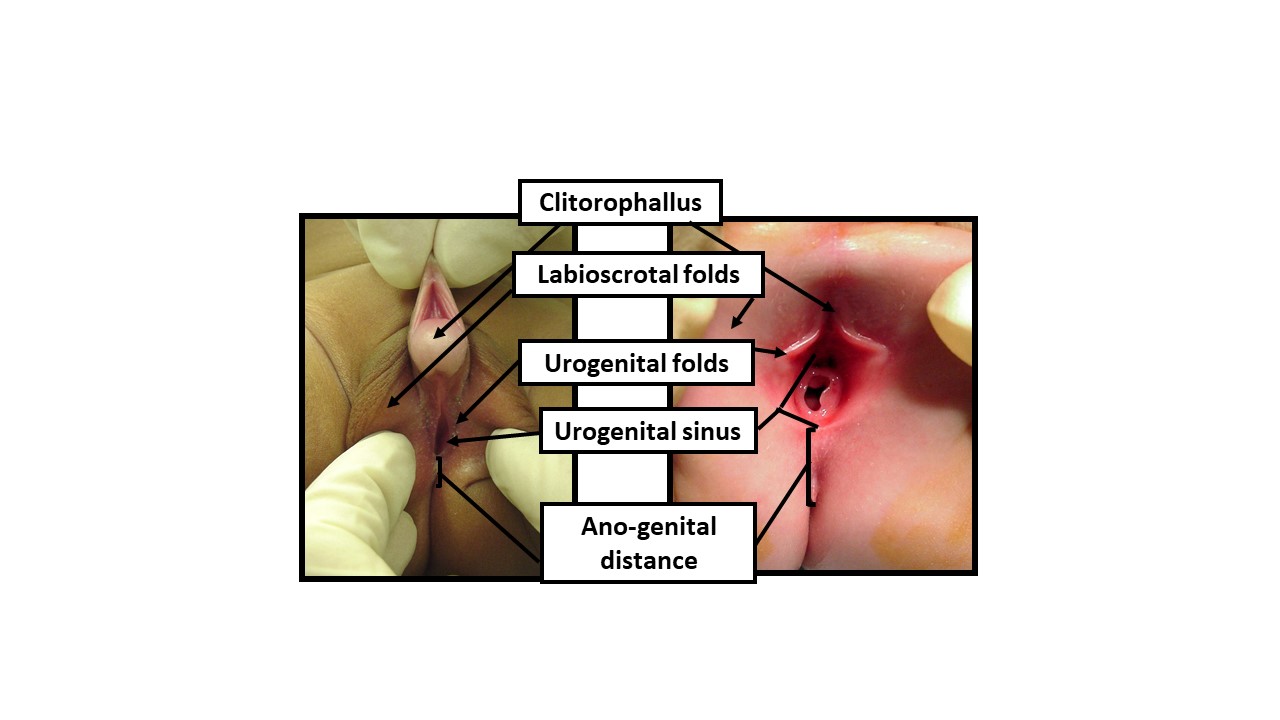
Figure 9 Description de l’examen du nouveau-né avec des termes anatomiques neutres du point de vue du genre. Lors de l’examen d’un nouveau-né présentant un aspect ambigu des organes génitaux, une terminologie neutre du point de vue du genre doit être utilisée jusqu’à ce que des données suffisantes permettent une prise de décision partagée avec les parents concernant la détermination du sexe d’élevage. L’examen doit consigner les signes suivants d’exposition aux androgènes : 1) Longueur de la structure clitoro-phallique, 2) Largeur du gland, 3) Nombre et position des orifices périnéaux, 4) Degré de fusion des plis labio-scrotaux, 5) Hyperpigmentation et rugosité des plis labio-scrotaux, 6) Distance anogénitale, 7) Localisation de toute gonade palpable
Une approche algorithmique fondée sur le nombre de gonades palpables et sur les résultats des examens endocriniens, génétiques et d’imagerie, comme indiqué ci‑dessus, peut aider efficacement à affiner le diagnostic différentiel. Chez le nouveau‑né, l’aspect médical le plus critique est d’écarter une hyperplasie congénitale des surrénales avec perte de sel mettant en jeu le pronostic vital. L’ensemble de l’équipe pluridisciplinaire décrite ci‑dessus joue un rôle durant cette période diagnostique critique pour aider la famille à intégrer les nouvelles informations et à prendre des décisions concernant le sexe d’élevage, la divulgation et la chirurgie.
Options de prise en charge chirurgicale
Les options chirurgicales pour les patients atteints de troubles du développement sexuel (DSD) sont hautement individualisées, et les décisions concernant le traitement chirurgical devraient idéalement être prises dans le cadre d’une équipe multidisciplinaire. Selon les besoins et les objectifs du patient, les interventions peuvent être diagnostiques, reconstructrices ou d’exérèse.
Procédures diagnostiques
Les procédures diagnostiques sont souvent entreprises lorsque l’anatomie ou le diagnostic demeurent incertains après une évaluation endocrinienne et génétique et une imagerie radiologique détaillée.
L’endoscopie permet une visualisation détaillée et des mesures de l’appareil urogénital. Par exemple, la longueur et la configuration d’un sinus urogénital peuvent être caractérisées. Les structures vaginales peuvent être évaluées quant à la profondeur, au nombre et à la présence ou à l’absence d’un col utérin.
La laparoscopie diagnostique facilite la visualisation des gonades et des structures müllériennes. Cette visualisation peut aider au diagnostic et fournir des informations pour la planification chirurgicale reconstructive future. Une biopsie gonadique peut être réalisée par voie laparoscopique ou par voie ouverte pour déterminer le type tissulaire (par exemple, si un DSD ovotesticulaire est suspecté), ou envoyée pour caryotypage si la présence de matériel du chromosome Y est suspectée mais n’a pas été identifiée au caryotype du sang périphérique. Figure 10 montre des images fixes laparoscopiques d’un patient atteint de MGD : le diagnostic de MGD a été suspecté cliniquement et confirmé par l’aspect macroscopique et histologique des gonades mis en évidence par la laparoscopie diagnostique et la biopsie.
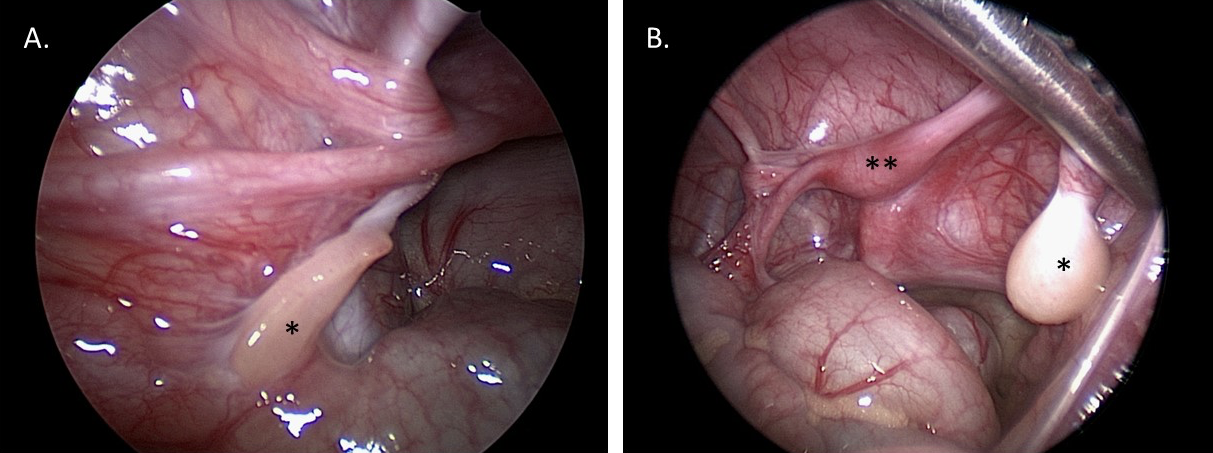
Figure 10 Images laparoscopiques diagnostiques pelviennes chez un patient âgé d’un an atteint de dysgénésie gonadique mixte. A. *Gonade en bandelette gauche. B. *Testicule dysgénétique droit. **Utérus.
Génitoplastie masculinisante
Chez les patients présentant un DSD, la génitoplastie de masculinisation implique le plus souvent la réparation d’un hypospadias proximal, souvent associée à une scrotoplastie pour corriger une transposition pénoscrotale et/ou une configuration bifide. Les modalités techniques de la réparation de l’hypospadias sont similaires à celles des patients sans DSD défini. La réparation de l’hypospadias chez les patients avec DSD nécessite généralement une procédure en deux temps afin d’obtenir la correction de la chordée et un méat urétral terminal. Il existe une controverse dans la littérature quant à savoir si les patients ayant un diagnostic de DSD connu présentent des résultats chirurgicaux de l’hypospadias plus défavorables par rapport aux patients avec un hypospadias proximal ne présentant pas de DSD spécifique.9,10,11
Génitoplastie féminisante
Les éléments comprennent la clitoroplastie (avec labioplastie) et la vaginoplastie. Il existe diverses approches, pouvant être adaptées en fonction de l’anatomie du patient et des objectifs. Un exemple de marquages chirurgicaux préopératoires pour un patient atteint de CAH ayant subi une mobilisation urogénitale partielle (PUM) et une labioplastie sans clitoroplastie est illustré dans Figure 11.
Clitoroplastie et labioplastie
Les procédures modernes de clitoroplastie les plus fréquemment rapportées impliquent une réduction clitoridienne avec préservation nerveuse.12 Le clitoris est dénudé et le tissu des corps (érectiles) est retiré, en évitant les faisceaux neurovasculaires dorsaux. L’approche de clitoroplastie de réduction épargnant la tunique albuginée a été introduite pour préserver davantage la vascularisation et la sensibilité glandulaires, et implique des incisions ventrales des corps et l’excision du tissu érectile, tout en préservant la tunique environnante. Le gland du clitoris est fréquemment préservé, enfoui proximalement et fixé sous le pubis. La réduction du gland peut être réalisée en excisant un coin de tissu ventral et en rapprochant les bords ventraux l’un de l’autre. Cependant, cette approche comporte un risque de diminution de la sensibilité glandulaire. Un clitoris enfoui peut souvent être obtenu grâce à une clitoroplastie associée à une labioplastie, de sorte que la réduction du gland devient moins fréquente.
Plusieurs techniques épargnant les corps caverneux ont émergé comme alternatives à la procédure de réduction clitoridienne décrite ci-dessus. À titre d’exemple, Pippi Salle a décrit une procédure de désassemblage des corps caverneux.13 Pour cette procédure, les corps caverneux sont complètement séparés du gland clitoridien et du paquet vasculo-nerveux, ainsi que l’un de l’autre. Les corps caverneux sont ensuite transposés individuellement par tunnelisation dans leurs grandes lèvres homolatérales respectives, et le gland clitoridien est repositionné proximalement, de manière similaire aux procédures de réduction clitoridienne. Cette « procédure de Pippi Salle » présente l’avantage théorique d’être réversible, puisqu’aucun tissu n’est retiré, bien que des tentatives d’inversion, ainsi que d’autres résultats à long terme, n’aient pas encore été rapportés.
La labioplastie constitue généralement l’étape finale au cours d’une procédure de clitoroplastie (avec ou sans vaginoplastie). Les objectifs sont de remodeler le capuchon clitoridien et les tissus de la hampe au-dessus du clitoris pour recouvrir le gland clitoridien, et d’avancer les berges cutanées vers l’arrière, latéralement au vestibule/orifice vaginal.
Vaginoplastie
L’approche de la vaginoplastie implique d’évaluer si la patiente présente un sinus urogénital avec une confluence basse (canal commun court avec séparation du vagin et de l’urètre près du périnée) ou une confluence haute (canal commun plus long et séparation plus haute.14
Une vaginoplastie par lambeau cutané périnéal (technique de Fortunoff) peut être suffisante chez les patientes ayant un sinus urogénital à confluence basse. Pour cette technique, on crée un lambeau en U postérieur (comme dans la Figure 11) La paroi postérieure du vagin est ensuite incisée proximalement par rapport à la confluence dysplasique, et le lambeau est positionné et fixé dans cette incision vaginale afin d’augmenter le calibre vaginal.
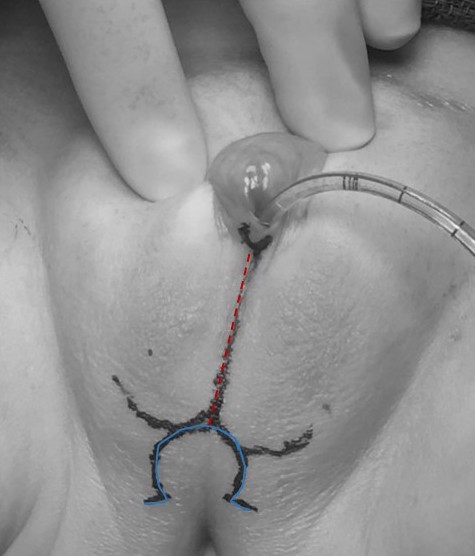
Figure 11 Marquage chirurgical préopératoire chez un patient présentant une hyperplasie congénitale des surrénales devant subir une mobilisation urogénitale partielle et une labioplastie sans clitoroplastie. Un cathéter est visible au niveau de l’orifice du sinus urogénital. Le marquage médian (rouge en pointillés) s’étend à travers les replis labioscrotaux fusionnés, et le marquage en forme d’oméga (bleu en trait plein) constituera le lambeau cutané postérieur pour la vaginoplastie.
Dans la mobilisation urogénitale totale (TUM) ou la mobilisation urogénitale partielle (PUM), l’urètre et le vagin sont mobilisés vers le périnée en un seul bloc.15 La différence entre les deux tient au fait que la dissection antérieure en arrière du pubis emporte le ligament pubo-urétral, comme illustré à la Figure 12. On peut craindre une incontinence urinaire avec une mobilisation plus étendue, mais cela n’a pas été démontré de manière définitive, et ce risque est probablement plus préoccupant si la distance de la confluence au col vésical est raccourcie. Lorsqu’elle est appliquée aux cas à confluence basse, la PUM amène généralement le vagin à une distance permettant son extériorisation ou la réalisation d’un pontage à l’aide d’un lambeau périnéal à pédicule postérieur (voir ci-dessus). La confluence haute signe une anatomie plus complexe, avec une distance plus grande pour atteindre le périnée, un rapport plus étroit entre le vagin et le col vésical, et souvent un vagin plus court. L’abaissement direct (pull-through) peut ne pas être avantageux à long terme. Dans les cas à confluence haute, même avec une mobilisation urogénitale, le vagin devra être séparé du sinus urogénital et abaissé jusqu’au périnée. Dans ce scénario, le sinus urogénital devient l’urètre. Le vagin atteint le périnée à l’aide de lambeaux cutanés périnéaux ou par l’utilisation, comme lambeau, d’un excès de tissu de sinus urogénital mobilisé.
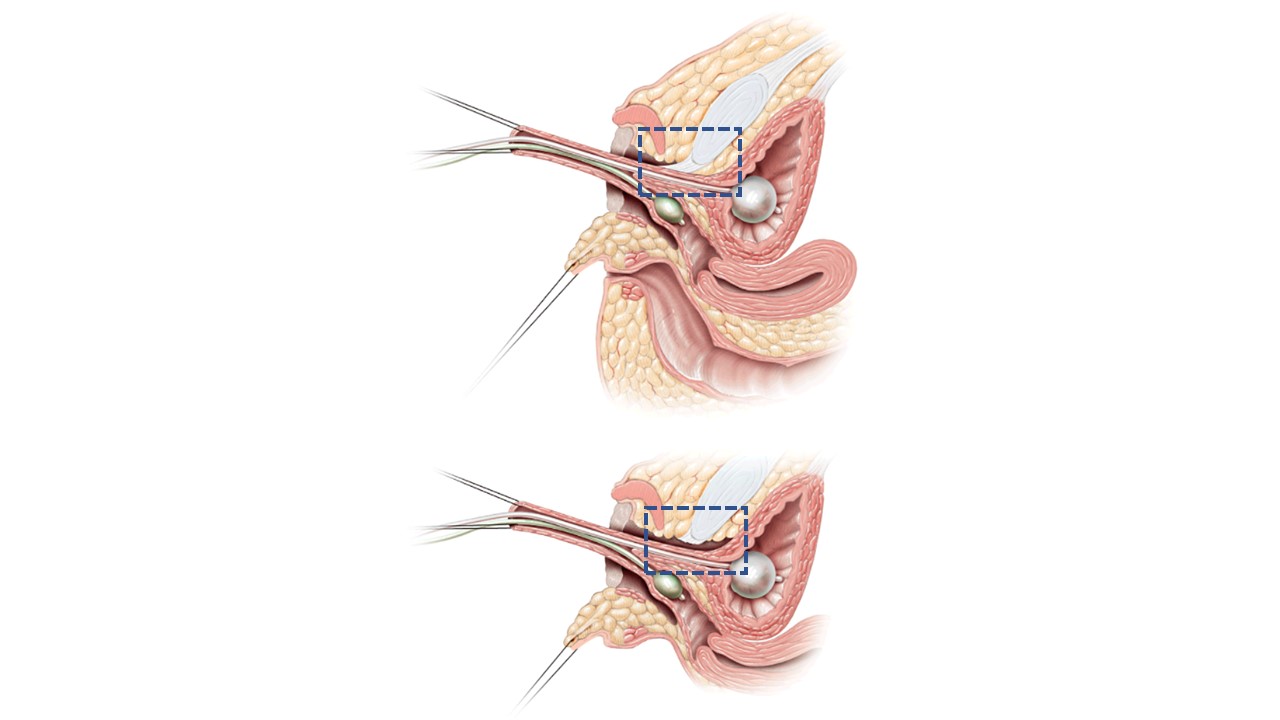
Figure 12 Images en coupe sagittale de la mobilisation urogénitale partielle vs totale. A. Mobilisation urogénitale partielle : l’urètre et le vagin sont avancés vers le périnée en bloc. Le ligament pubo-urétral (encadré en pointillés bleus) est laissé intact. B. Mobilisation urogénitale totale : l’urètre et le vagin sont avancés vers le périnée en bloc. Le ligament pubo-urétral (encadré en pointillés bleus) est sectionné. Adapté de : Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int 2008; 102 (9): 1182–1197. DOI: 10.1111/j.1464-410x.2008.08091.x. (autorisation accordée le 2/1/2022)
Chez les patients qui naissent avec des structures müllériennes absentes ou mal développées, la vaginoplastie consiste en la création d’un néovagin par dissection à bout mousse dans l’espace rétrovésical entre la vessie et le rectum, puis à tapisser le nouvel espace potentiel soit avec du tissu autologue (p. ex., greffe cutanée, greffe de muqueuse buccale, lambeau péritonéal), soit avec d’autres greffes biologiques (p. ex., sous-muqueuse de l’intestin grêle [SIS]). Ces techniques peuvent également être utilisées en complément des procédures décrites ci-dessus, ou dans un contexte de reprise chirurgicale.
Dilatations vaginales
Certains patients atteints de DSD (p. ex., AIS complète) peuvent obtenir un vagin permettant une activité sexuelle confortable et plaisante grâce à des dilatations vaginales régulières, sans avoir besoin d’une vaginoplastie chirurgicale. Tous les patients qui subissent une vaginoplastie chirurgicale doivent également effectuer des dilatations vaginales pendant la cicatrisation après l’opération, et jusqu’à ce qu’ils aient des rapports vaginaux de manière régulière.
Gonadectomie et prise en charge gonadique
Le paradigme de la prise en charge gonadique chez les patients présentant un DSD a évolué au fil du temps. Traditionnellement, chez les patients avec des DSD comportant un risque accru de formation tumorale, il était conseillé de subir une gonadectomie. L’infertilité était présumée. Avec le temps, il est apparu qu’il existe un large éventail de risques tumoraux et que les tumeurs qui se forment sont fréquemment des gonadoblastomes (non malins).16,17 Les dysgerminomes (tumeurs malignes des cellules germinales) sont possibles, mais extrêmement rares avant la puberté. Il peut également exister un potentiel de fertilité non traditionnel chez certaines personnes auparavant considérées comme infertiles.18 Ainsi, les recommandations de prise en charge des gonades deviennent plus individualisées, en tenant compte du diagnostic du patient (et donc du risque tumoral prédit), de l’âge et du potentiel de fonction gonadique endogène (tant hormonale que reproductive). Pour les patients qui optent pour la gonadectomie, la cryoconservation de tissu gonadique est une option émergente qui pourrait permettre une fertilité biologique, sous réserve que les progrès attendus des technologies de procréation assistée se concrétisent.19 Aucun protocole de surveillance fondé sur des preuves n’est encore disponible pour les patients qui choisissent de conserver leurs gonades; l’établissement des auteurs recommande généralement une échographie pelvienne tous les 6–12 mois. Tableau 4 présente des stratégies de recommandation pour la prise en charge gonadique dans plusieurs diagnostics de DSD.
Tableau 4 Options de prise en charge gonadique selon le diagnostic. * envisager la cryoconservation expérimentale de tissu gonadique.
| Diagnostic | Risque tumoral estimé | Cellules germinales attendues ? | Puberté attendue ? | Options de prise en charge des gonades |
|---|---|---|---|---|
| Dysgénésie gonadique complète 46, XY (syndrome de Swyer) | Élevé – Jusqu’à 35 % |
Non (éventuellement des cellules germinales précurseurs) |
Non | -Gonadectomie* |
| Syndrome d’insensibilité partielle aux androgènes | Élevé – Jusqu’à 50 % selon la localisation des gonades |
Oui Spermatozoïdes possibles |
Oui, variable | -Orchidopexie, envisager une biopsie -Observation avec échographies -Gonadectomie* |
| Syndrome de Turner + chromosome Y | Modéré – ~10-30 % |
Peut-être – Ovocytes possibles | Possible | -Observation avec échographies -Gonadectomie* (standard, faible niveau de preuve) |
| Syndrome d’insensibilité complète aux androgènes | Faible – ~2-10 % |
Oui – Précurseurs des spermatozoïdes |
Oui – en raison de l’aromatisation de la testostérone en œstrogènes | -Gonadectomie post-pubertaire* -Observation avec échographies, IRM |
Prise en charge des structures müllériennes
Chez les patients élevés comme garçons et présentant des structures müllériennes persistantes (c.-à-d., un utricule volumineux), celles-ci sont en général laissées en place tant qu’elles ne deviennent pas symptomatiques. L’excision expose à un risque de lésion des structures du canal déférent (et possiblement des nerfs pelviens), et les patients sont fréquemment asymptomatiques. La réparation de l’hypospadias est sûre, et l’excision, même de structures utriculaires volumineuses, n’est pas obligatoire avant la réparation de l’hypospadias. En cas de symptômes tels que des infections ou une hématurie cyclique gênante, une excision chirurgicale peut être envisagée par voie ouverte, laparoscopique ou robot-assistée. Une prise en charge hormonale avec suppression menstruelle peut également être envisagée chez les patients présentant des douleurs cycliques dues à un reliquat müllérien obstrué, ou une hématurie cyclique gênante.
Conclusions
Les DSD englobent un large éventail d’affections résultant d’un développement chromosomique, gonadique ou anatomique anormal. Il existe une large gamme de phénotypes, même au sein de diagnostics et de catégories de DSD spécifiques. Une approche multidisciplinaire de l’évaluation et du suivi à long terme est recommandée, dans la mesure du possible.
Points clés
- La terminologie DSD acceptée sur le plan médical n’est pas préférée par toutes les personnes concernées ni par leurs familles. Lors des consultations cliniques individuelles, demandez aux patients et aux familles quels termes ils préfèrent utiliser pour parler du diagnostic et de l’anatomie.
- Les affections DSD surviennent en raison de chromosomes, de gonades ou d’une action hormonale atypiques. Réfléchir au degré d’anomalie et déterminer si le défaut identifié est complet ou partiel peut aider à prédire le phénotype du patient.
- Le nombre de gonades palpables constitue la base d’un diagnostic différentiel de DSD. Le diagnostic différentiel est ensuite affiné par le caryotype, les bilans hormonaux et l’échographie pelvienne.
- N’oubliez pas de féliciter les nouveaux parents qui ont un nourrisson avec un DSD suspecté—cette étape cruciale est souvent oubliée.
- La CAH est la forme la plus fréquente de DSD 46, XX. Les caractéristiques cliniques incluent des organes génitaux externes virilisés avec des gonades non palpables. Une perte de sel mettant en jeu le pronostic vital peut survenir à 1 semaine de vie.
- Le DSD ovotesticulaire peut survenir chez un patient avec n’importe quel caryotype.
- Un patient avec un caryotype 45, X/46, XY peut avoir des organes génitaux externes de type féminin, ambigus ou de type masculin. Tout patient avec ce caryotype peut présenter des manifestations systémiques du syndrome de Turner.
Lectures recommandées
- Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
- Kaefer M, Diamond D, Hendren WH. The Incidence Of Intersexuality In Children With Cryptorchidism And Hypospadias: Stratification Based On Gonadal Palpability And Meatal Position. J Urol 1999; 162 (3 Part 2): 1006–1007. DOI: 10.1016/s0022-5347(01)68049-2.
- Kim S, Rosoklija I, Johnson EK. Surgical, Patient, and Parental Considerations in the Management of Children with Differences of Sex Development. Curr Pediatr Rep 2018; 6 (3): 209–219. DOI: 10.1007/s40124-018-0177-4.
- Mouriquand PD, Gorduza DB, Gay CL. Re: “Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how?” J Pediatr Urol 2016; 12 (6): 442–443. DOI: 10.1016/j.jpurol.2016.07.013.
- Nixon R, Cerqueira V, Kyriakou A, Lucas-Herald A, McNeilly J, McMillan M, et al.. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod 2017; 32 (10): 2130–2137. DOI: 10.1093/humrep/dex280.
- Van Batavia JP, Kolon TF. Fertility in disorders of sex development: A review. J Pediatr Urol 2016; 12 (6): 418–425. DOI: 10.1016/j.jpurol.2016.09.015.
Références
- Lee PA, Houk CP, Ahmed SF. Consensus Statement on Management of Intersex Disorders. Pediatric Clinical Practice Guidelines &Amp; Policies 2006; 118: 1317–1317. DOI: 10.1542/9781610021494-part06-consensus_statement2.
- Johnson EK, Rosoklija I, Finlayson C. Faculty Opinions recommendation of Attitudes towards "disorders of sex development" nomenclature among affected individuals. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2017; 13: 608. DOI: 10.3410/f.727655647.793533819.
- D’Oro A, Rosoklija I, Jacobson DL, Finlayson C, Chen D, Tu DD, et al.. Patient and Caregiver Attitudes toward Disorders of Sex Development Nomenclature. J Urol 2020; 204 (4): 835–842. DOI: 10.1097/ju.0000000000001076.
- Davies JH, Knight EJ, Savage A, Brown J, Malone PS. Evaluation of terminology used to describe disorders of sex development. J Pediatr Urol 2011; 7 (4): 412–415. DOI: 10.1016/j.jpurol.2010.07.004.
- Lin-Su K, Lekarev O, Poppas DP, Vogiatzi MG. Congenital adrenal hyperplasia patient perception of ‘disorders of sex development’ nomenclature. Int J Pediatr Endocrinol 2015; 2015 (1): 9. DOI: 10.1186/s13633-015-0004-4.
- Snodgrass W, Macedo A, Hoebeke P, Mouriquand PDE. Hypospadias dilemmas: A round table. J Pediatr Urol 2011; 7 (2): 145–157. DOI: 10.1016/j.jpurol.2010.11.009.
- Wu Q, Wang C, Shi H, Kong X, Ren S, Jiang M. The Clinical Manifestation and Genetic Evaluation in Patients with 45,X/46,XY Mosaicism. Sex Dev 2017; 11 (2): 64–69. DOI: 10.1159/000455260.
- Das DV, Jabbar PK. Clinical and Reproductive Characteristics of Patients with Mixed Gonadal Dysgenesis (45,X/46, XY). J Obstet Gynaecol India 2021; 71 (4): 399–405. DOI: 10.1007/s13224-021-01448-3.
- Saltzman AF, Carrasco A, Colvin A, Campbell JB, Vemulakonda VM, Wilcox D. Patients with disorders of sex development and proximal hypospadias are at high risk for reoperation. World J Urol 2018; 36 (12): 2051–2058. DOI: 10.1007/s00345-018-2350-3.
- Ochi T, Ishiyama A, Yazaki Y, Murakami H, Takeda M, Seo S, et al.. Surgical management of hypospadias in cases with concomitant disorders of sex development. Pediatr Surg Int 2019; 35 (5): 611–617. DOI: 10.1007/s00383-019-04457-6.
- Palmer BW, Reiner W, Kropp BP. Proximal Hypospadias Repair Outcomes in Patients with a Specific Disorder of Sexual Development Diagnosis. Adv Urol 2012; 2012: 1–4. DOI: 10.1155/2012/708301.
- Kaefer M, Rink RC. Treatment of the Enlarged Clitoris. Front Pediatr 2017; 5. DOI: 10.3389/fped.2017.00125.
- Pippi Salle JL, Braga LP, Macedo N, Rosito N, Bagli D. Corporeal Sparing Dismembered Clitoroplasty: An Alternative Technique for Feminizing Genitoplasty. J Urol 2007; 178 (4s): 1796–1801. DOI: 10.1016/j.juro.2007.03.167.
- Guarino N, Scommegna S, Majore S, Rapone AM, Ungaro L, Morrone A, et al.. Vaginoplasty for Disorders of Sex Development. Front Endocrinol (Lausanne) 2013; 4: 29, DOI: 10.3389/fendo.2013.00029.
- Rink RC, Cain MP. Urogenital mobilization for urogenital sinus repair. BJU Int 2008; 102 (9): 1182–1197. DOI: 10.1111/j.1464-410x.2008.08091.x.
- Looijenga LHJ, Hersmus R, Oosterhuis JW, Cools M, Drop SLS, Wolffenbuttel KP. Tumor risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab 2007; 21 (3): 480–495. DOI: 10.1016/j.beem.2007.05.001.
- Looijenga LHJ, Hersmus R, Leeuw BHCGM de, Stoop H, Cools M, Oosterhuis JW, et al.. Gonadal tumours and DSD. Best Pract Res Clin Endocrinol Metab 2010; 24 (2): 291–310. DOI: 10.1016/j.beem.2009.10.002.
- Finlayson C, Fritsch MK, Johnson EK, Rosoklija I, Gosiengfiao Y, Yerkes E, et al.. Presence of Germ Cells in Disorders of Sex Development: Implications for Fertility Potential and Preservation. J Urol 2017; 197 (3 Part 2): 937–943. DOI: 10.1016/j.juro.2016.08.108.
- Harris CJ, Corkum KS, Finlayson C, Rowell EE, Laronda MM, Reimann MB, et al.. Establishing an Institutional Gonadal Tissue Cryopreservation Protocol for Patients with Differences of Sex Development. J Urol 2020; 204 (5): 1054–1061. DOI: 10.1097/ju.0000000000001128.
- Kaefer M, Diamond D, Hendren WH. The Incidence Of Intersexuality In Children With Cryptorchidism And Hypospadias: Stratification Based On Gonadal Palpability And Meatal Position. J Urol 1999; 162 (3 Part 2): 1006–1007. DOI: 10.1016/s0022-5347(01)68049-2.
- Kim S, Rosoklija I, Johnson EK. Surgical, Patient, and Parental Considerations in the Management of Children with Differences of Sex Development. Curr Pediatr Rep 2018; 6 (3): 209–219. DOI: 10.1007/s40124-018-0177-4.
- Mouriquand PD, Gorduza DB, Gay CL. Re: “Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how?” J Pediatr Urol 2016; 12 (6): 442–443. DOI: 10.1016/j.jpurol.2016.07.013.
- Nixon R, Cerqueira V, Kyriakou A, Lucas-Herald A, McNeilly J, McMillan M, et al.. Prevalence of endocrine and genetic abnormalities in boys evaluated systematically for a disorder of sex development. Hum Reprod 2017; 32 (10): 2130–2137. DOI: 10.1093/humrep/dex280.
- Van Batavia JP, Kolon TF. Fertility in disorders of sex development: A review. J Pediatr Urol 2016; 12 (6): 418–425. DOI: 10.1016/j.jpurol.2016.09.015.
Dernière mise à jour: 2025-09-22 07:59