
13: Maladies kystiques du rein
Ce chapitre prendra environ 24 minutes de lecture.
Introduction
Les maladies kystiques rénales constituent une vaste catégorie d’affections rénales sporadiques et génétiques, congénitales ou acquises, pouvant aller de la présence d’un kyste unique à de multiples kystes touchant un ou les deux reins (voir les classifications héréditaires dans le Tableau 1 ci-dessous).1 Selon le Comité de 1987 sur la classification, la nomenclature et la terminologie, la classification des maladies kystiques rénales est principalement déterminée par l’étiologie génétique ou non génétique.2 Les maladies kystiques rénales sont globalement considérées comme des affections rares ; toutefois, elles peuvent être associées à des conséquences graves, telles qu’une maladie rénale chronique (CKD), une insuffisance rénale ou une maladie hépatique. Malgré leur rareté, la polykystose rénale autosomique dominante (ADPKD) est l’une des affections héréditaires humaines les plus courantes. La possibilité de complications graves liées aux maladies kystiques rénales dans la population pédiatrique, en particulier la maladie rénale chronique (CKD), confère une importance certaine au diagnostic précoce et à des modalités thérapeutiques efficaces.
Tableau 1 Classifications héréditaires des maladies kystiques rénales.1
| Héréditaire | Non héréditaire |
|---|---|
| Polykystose rénale autosomique récessive (infantile) | Rein multikystique (rein dysplasique multikystique) |
| Polykystose rénale autosomique dominante (adulte) | Kyste multiloculaire bénin (néphrome kystique) |
| Complexe de néphronophtise juvénile et de maladie kystique médullaire | Kystes simples |
| Néphronophtise juvénile (autosomique récessive) | Rein en éponge médullaire |
| Maladie kystique médullaire (autosomique dominante) | Maladie rénale glomérulo-kystique sporadique |
| Néphrose congénitale (syndrome néphrotique familial) (autosomique récessive) | Maladie kystique rénale acquise |
| Maladie glomérulo-kystique hypoplasique familiale (autosomique dominante) | Diverticule caliciel (kyste pyélogène) |
| Syndromes de malformations multiples avec kystes rénaux (p. ex., sclérose tubéreuse, maladie de von Hippel-Lindau) |
Embryologie
Les kystes surviennent généralement secondairement à des déséquilibres de sécrétion, d’absorption et électrolytiques au niveau des cellules épithéliales et peuvent se former partout dans le système tubulaire rénal. Les recherches récentes sur les maladies rénales kystiques héréditaires suggèrent que les gènes en cause impliquent le cil primaire des cellules épithéliales rénales, une voie commune potentielle pour ces affections.3 Le terme “ciliopathie” désigne cette voie commune. Les cils ont pour fonction de moduler la prolifération et la différenciation des cellules tubulaires, et leur dysfonction peut entraîner une expansion inappropriée des tubules et la formation de kystes.4
Ce chapitre traitera de l’épidémiologie, de la physiopathologie, de l’évaluation et du traitement de diverses maladies rénales kystiques héréditaires et non héréditaires, notamment l’ADPKD, la polykystose rénale autosomique récessive (ARPKD), les kystes rénaux isolés, les diverticules caliciels, le rein multikystique dysplasique (MCDK), le rein médullaire en éponge, la néphronophthisie, les kystes multiloculaires (néphrome kystique multiloculaire), les maladies rénales kystiques acquises et les syndromes associés aux maladies rénales kystiques.
Polykystose rénale autosomique dominante
L’ADPKD est relativement fréquente, avec une incidence de 1 pour 400 à 1 000 naissances vivantes, se manifestant par une dilatation kystique du néphron et d’autres anomalies extrarénales, telles qu’un anévrisme intracrânien.5 La gravité varie selon les patients et tend à s’aggraver avec le temps; ainsi, de nombreux patients pédiatriques et même certains adultes d’âge moyen peuvent être asymptomatiques. Bien que rare, une forme sévère pendant l’enfance peut augurer d’une morbidité et d’une mortalité importantes.6 En raison des antécédents familiaux ou de découvertes fortuites à l’imagerie, un plus grand nombre d’enfants asymptomatiques sont identifiés comme atteints d’ADPKD.
Épidémiologie
Parmi les personnes atteintes d’ADPKD, environ 96 % présenteront des signes cliniques de la maladie avant l’âge de 90 ans.7 Bien que la majorité des cas soient découverts chez des patients d’âge moyen (30–40 ans) et que l’insuffisance rénale avant 40 ans soit rare, la maladie a été décrite dès la période néonatale. Lorsqu’elle est décrite chez les nouveau-nés, l’ADPKD est considérée comme plus agressive.8 Malgré un mode de transmission autosomique dominant avec une pénétrance théorique de 100 %, 10 % des cas peuvent survenir de façon sporadique.1
Physiopathologie
L’ADPKD est héritée selon un mode autosomique dominant et les principales mutations génétiques surviennent dans PKD1 et PKD2 sur le chromosome 16 et le chromosome 4, respectivement.5 Le produit génique est une protéine transmembranaire (polycystine 1 et polycystine 2, respectivement) localisée au niveau des cils primaires.9 Les mutations de PKD1 sur le chromosome 16 représentent la majorité des cas et sont associées à une sévérité accrue, tandis que la plupart des autres cas sont dus à des mutations de PKD2.10
Un fonctionnement anormal de la polycystine 1 ou de la polycystine 2 entraîne une dérégulation des signaux des voies prolifératives telles que cAMP, ERK et mTOR. Les cils pourraient jouer un rôle organisationnel dans la transduction de ces signaux, la dérégulation entraînant la formation de kystes.11
Évaluation et diagnostic
En tant qu’examen d’imagerie coût-efficace et peu invasif, l’échographie rénale est fréquemment utilisée pour identifier l’ADPKD. Chez les sujets de moins de 30 ans, les critères diagnostiques échographiques comprennent la présence d’au moins deux kystes unilatéraux ou bilatéraux, un plus grand nombre de kystes étant nécessaire pour suggérer le diagnostic avec l’augmentation de l’âge.12 Cependant, l’ADPKD et l’ARPKD peuvent être difficiles à distinguer dans la population pédiatrique, ce qui rend la présence d’antécédents familiaux positifs cruciale pour le diagnostic.13 En l’absence d’antécédents familiaux, des tests génétiques peuvent être réalisés. D’autres facteurs importants à considérer, lorsque les antécédents familiaux sont négatifs, comprennent une augmentation bilatérale du volume rénal, trois kystes hépatiques ou plus, un anévrysme d’une artère cérébrale, et des kystes solitaires d’autres organes (arachnoïde, glande pinéale, pancréas ou rate).1,14 L’imagerie ou la biopsie du foie peut également aider à différencier l’ADPKD et l’ARPKD, car la fibrose hépatique est toujours présente dans l’ARPKD, mais rare dans l’ADPKD.5 Enfin, l’anatomopathologie rénale (biopsie) peut être utile, car l’ADPKD peut intéresser l’ensemble du tubule, y compris le glomérule; en revanche, l’ARPKD ne présente pas de kystes glomérulaires.5
Options de traitement, résultats et complications
Il n’existe actuellement aucun traitement curatif de la PKRAD. Les objectifs du traitement incluent de retarder l’apparition de l’insuffisance rénale terminale (IRT) et de réduire la charge des complications de la PKRAD liées au déclin de la fonction rénale, à la maladie cardiaque et à l’hémorragie intracrânienne. Les complications sont le plus significativement modulées par la prise en charge de l’hypertension artérielle.1 Par conséquent, un contrôle rigoureux de la pression artérielle est recommandé et les inhibiteurs de l’enzyme de conversion de l’angiotensine (IEC) ainsi que les antagonistes des récepteurs de l’angiotensine II (ARA II) sont des choix courants; cependant, il n’existe pas de consensus sur l’antihypertenseur idéal.15,16
Les thérapies émergentes en cours d’investigation visent à limiter la croissance des kystes en réduisant l’AMPc au moyen d’inhibiteurs de mTOR, de la somatostatine, d’inhibiteurs de tyrosine kinase, d’inhibiteurs de la kinase Src, d’inhibiteurs de MEK et d’inhibiteurs des kinases dépendantes des cyclines.
Maladie rénale polykystique autosomique récessive
L’ARPKD se caractérise par une augmentation rapide du volume des deux reins chez les nourrissons due à la formation de kystes au sein du système des tubes collecteurs. Elle peut être associée à une fibrose hépatique congénitale pouvant entraîner une hypertension portale. Une présentation plus précoce d’ARPKD est souvent associée à une sévérité accrue.1 Ses effets multisystémiques peuvent nécessiter une prise en charge multidisciplinaire.
Épidémiologie
Comme l’ARPKD est moins fréquente que l’ADPKD, il existe moins de données épidémiologiques. L’incidence rapportée est de 1 sur 26,500 naissances vivantes, mais peut varier de 1 sur 10,000 à 50,000 naissances vivantes.1,17 Cependant, elle peut être plus fréquente dans les populations isolées ou consanguines.
Pathogénie
L’ARPKD est héritée selon un mode autosomique récessif classique, les parents hétérozygotes non atteints ayant un risque de 25 % d’avoir un enfant atteint. Elle est due à une mutation du gène polycystic kidney and hepatic disease 1 (PKHD1) sur le chromosome 6, qui code la fibrocystine (également appelée polyductine), une protéine identifiée dans le système tubulaire rénal (canal collecteur et branche ascendante épaisse), dans les cellules épithéliales du canal biliaire hépatique, et dans les cils primaires apicaux.5,18,19
Un patient typique peut se présenter en période néonatale avec des antécédents d’oligoamnios prénatal, de reins volumineux, et une séquence de “Potter” avec hypoplasie pulmonaire et décès périnatal dans environ 30 % des nouveau-nés atteints.20,21 Les reins volumineux apparaissent hyperéchogènes avec une dilatation fusiforme des canaux collecteurs, et l’évolution vers une insuffisance rénale terminale (IRT) peut survenir à des âges variables. L’atteinte d’autres systèmes d’organes peut se manifester par des voies biliaires dilatées, une fibrose hépatique congénitale, une hypertension portale et une dysfonction neurocognitive.22
Évaluation et diagnostic
L’échographie fœtale et néonatale peut montrer des reins bilatéraux, très augmentés de taille et diffusément échogènes en raison d’une abondance de microkystes.1 La présence de macrokystes (> 10 mm) peut indiquer d’autres ciliopathies telles que MCDK ou ADPKD, et l’IRM peut aider à évaluer plus avant les anomalies dans un contexte d’oligoamnios sévère.13 Un diagnostic précis dépend de résultats d’imagerie évocateurs, de la présence d’antécédents familiaux avec mode de transmission récessif, d’une biopsie hépatique montrant des lésions périportales (présentes dans l’ARPKD et rares dans l’ADPKD), et de l’absence de manifestations extra-rénales associées à d’autres maladies kystiques rénales.1
Options de traitement, résultats et complications
Comme pour l’ADPKD, il n’existe pas de traitement curatif pour l’ARPKD, de sorte que de nombreuses options thérapeutiques visent la prise en charge des symptômes ou la palliation liées à l’hypertension, à l’insuffisance cardiaque congestive, ainsi qu’à l’insuffisance rénale et hépatique. Un traitement agressif, tel qu’une néphrectomie unilatérale ou bilatérale, peut être indiqué chez les personnes sévèrement atteintes qui présentent une altération respiratoire ou nutritionnelle due à l’effet de masse de reins augmentés de volume. Des thérapies décompressives peuvent être mises en œuvre pour la prise en charge de l’hypertension portale. L’hémodialyse et la transplantation rénale sont souvent envisagées.1
Kystes rénaux isolés
Les kystes rénaux solitaires et multiples sont les lésions les plus fréquentes du rein de l’adulte et du sujet âgé, mais ils sont rares chez les enfants et les nourrissons.4 Ils ont été identifiés en période prénatale, bien que les lésions prénatales se résolvent fréquemment au cours de la grossesse.23
Épidémiologie
L’échographie prénatale a montré une prévalence des kystes rénaux de 0,09 %.23 L’incidence des kystes simples, de la naissance à 18 ans, est en moyenne de 0,22 %, avec un intervalle de 0,1 % à 0,45 %.24 L’incidence augmente chez les adultes et avec l’âge.
Pathogénie
La taille est très variable; toutefois, la plupart des kystes mesurent moins de 2 cm et sont souvent corticaux, altérant le contour rénal, mais ils peuvent aussi être corticaux profonds ou d’apparence médullaire, sans continuité avec le bassinet rénal.1 Ils n’altèrent généralement pas la fonction rénale, mais peuvent occasionnellement provoquer des douleurs lorsqu’un kyste comprime les structures adjacentes ou obstrue le système collecteur.5
Évaluation et diagnostic
L’identification est généralement fortuite et le diagnostic peut être posé en toute sécurité par échographie lorsque les critères suivants sont réunis : 1) kyste à contenu anéchogène, 2) paroi fine, nettement délimitée, à contours bien distincts, 3) transmission adéquate des ondes ultrasonores avec renforcement acoustique postérieur, 4) forme sphérique ou ovoïde.1 Si ces critères ne sont pas remplis ou lorsque l’échographie ne fournit pas une évaluation satisfaisante, comme dans le cas de kystes plus complexes, le scanner (TDM) peut apporter davantage de détails anatomiques. À défaut, une IRM ou une ponction-aspiration à l’aiguille peut être indiquée.25
Les kystes simples ne communiquent pas avec le bassinet rénal; toutefois, certains kystes peuvent apparaître à proximité immédiate. La suspicion d’une communication peut être évaluée par une imagerie en coupes avec contraste intraveineux et phase tardive, telle qu’une uro-TDM ou une uro-IRM. De plus, la ponction kystique et les analyses de laboratoire peuvent aider à identifier une communication avec le bassinet rénal. Si non communicant, l’azote uréique sanguin et la créatinine seront comparables aux valeurs du patient.5,26,27 Un kyste isolé peut également faire suspecter une ADPKD en cas d’antécédents familiaux positifs.28 Historiquement, la classification de Bosniak des kystes rénaux n’a pas été bien validée chez l’enfant; toutefois, une étude récente conclut qu’un système Bosniak modifié fournit une stratification du risque raisonnable et a montré que les lésions avec un score mBosniak de 1 ou 2 sont souvent bénignes, tandis que les scores 3 ou 4 présentaient dans 90 % des cas une pathologie intermédiaire ou maligne.29
Options de traitement et leurs résultats
Les kystes symptomatiques peuvent être drainés, mais ils risquent de récidiver en l’absence d’un agent sclérosant.5 Une autre option thérapeutique consiste en une intervention chirurgicale avec décortication kystique, qui se prête à une approche laparoscopique mini-invasive, en particulier pour les kystes relativement superficiels et/ou exophytiques.
Le suivi peut consister en une échographie pour surveiller des signes évocateurs de malignité. Les kystes stables qui n’ont pas augmenté de taille pendant deux ans peuvent ne nécessiter aucun suivi supplémentaire.29,30,31,32,33 Une intervention peut ne pas être indiquée après qu’il a été démontré que la lésion n’est pas maligne.
Diverticules caliciels
Les diverticules caliciels (DC) sont de rares évaginations du calice dans le parenchyme rénal. Ils sont classés en deux groupes : le type 1, plus fréquent, est adjacent à un calice supérieur ou inférieur, tandis que le type 2 est plus volumineux, communique avec le bassinet rénal et est plus souvent symptomatique.34
Épidémiologie
Il a été rapporté que le CD est retrouvé chez 0,2 % à 0,6 % des enfants ayant eu une urographie intraveineuse.13
Pathogénie
La pathogénie du CD est largement inconnue et n’est pas considérée comme une maladie héréditaire d’origine génétique. Cependant, certaines théories incluent des mutations géniques induisant des syndromes qui affectent le rein, ou, alternativement, un défaut de régression du bourgeon urétéral ou des causes obstructives.13 Le kyste est tapissé d’un épithélium transitionnel et communique avec le système collecteur, généralement par un calice ou un infundibulum rétréci ou sténosé, et plus rarement par le bassinet rénal.35
Évaluation et diagnostic
Les patients peuvent être asymptomatiques ou plutôt se présenter avec une hématurie, une douleur, une infection urinaire ou une lithiase rénale ; le signe de présentation le plus fréquent chez l’enfant est l’infection urinaire fébrile.35 Les diverticules asymptomatiques sont généralement découverts lors d’examens d’imagerie. Bien que l’échographie puisse identifier initialement le diverticule caliciel, afin de distinguer un kyste simple d’un diverticule caliciel, l’examen d’imagerie le plus précis est l’imagerie en coupes avec contraste IV et phase tardive.35,36,37 Figure 1 et Figure 2 montrent des signes compatibles avec un CD à l’urographie par IRM. Une étude récente comparant l’échographie à l’imagerie en coupes a montré que l’échographie rénale ciblée avait une sensibilité de 40% et une spécificité de 100% respectivement, tandis que l’imagerie en coupes (en particulier l’urographie par IRM à liquide statique) avait une sensibilité de 100% et une spécificité de 91.6% respectivement.37 La douleur, l’infection, la formation d’un abcès, l’urosepsie et des calculs rénaux symptomatiques peuvent tous constituer des indications possibles d’une prise en charge chirurgicale.
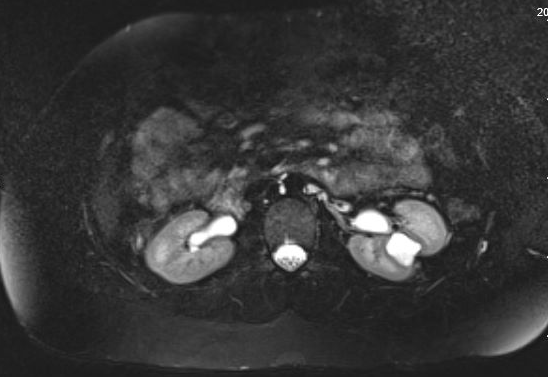
Figure 1 Urographie par IRM montrant l’opacification d’un diverticule caliciel dans la région interpolaire du rein gauche, avec amincissement sus-jacent du parenchyme rénal.
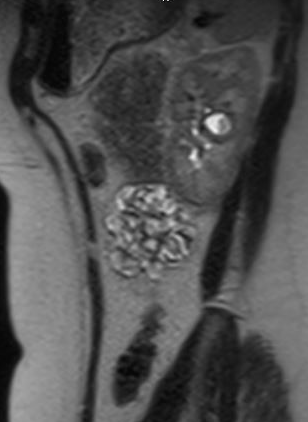
Figure 2 Vue sagittale d’une urographie par IRM chez le même patient, montrant l’opacification du diverticule caliciel par le produit de contraste.
Options thérapeutiques, leurs résultats et complications
Les patients asymptomatiques ne nécessitent pas de traitement, mais peuvent bénéficier d’une surveillance échographique.1 Ceux qui présentent des indications de prise en charge chirurgicale, comme mentionné ci-dessus, peuvent bénéficier d’une marsupialisation laparoscopique du diverticule avec fulguration du revêtement épithélial.38,39,40 D’autres procédures de prise en charge possibles peuvent inclure l’urétéroscopie ou la néphrolithotomie percutanée mini-invasive (en cas de calculs),41 l’ablation urétéroscopique de la cavité,39 ou l’ablation et la fulguration percutanées.42 Malgré la variété des possibilités thérapeutiques, l’intervention chirurgicale pour la prise en charge du CD peut être difficile. Dans le but de minimiser la morbidité, la prise en charge débute généralement par les options les moins invasives, telles que les approches endoscopiques ou percutanées. Il peut exister un taux élevé de récidive des symptômes,42 auquel cas l’intervention peut être intensifiée vers des approches laparoscopiques/robotiques et même des approches ouvertes, pouvant aller jusqu’à une néphrectomie partielle ou une néphrectomie totale. Par conséquent, une information et un accompagnement approfondis des patients sont nécessaires lorsqu’on envisage une intervention chirurgicale pour le CD.
Rein dysplasique multikystique
Le rein dysplasique multikystique (MCDK) est une malformation congénitale fréquemment diagnostiquée pendant l’enfance ou par échographie prénatale, caractérisée par de nombreux kystes unilatéraux non communicants et une différenciation métanéphrique anormale (présence de cartilage, de mésenchyme indifférencié et de canalicules collecteurs immatures), souvent dans un contexte d’uropathie obstructive.4,13,43 L’atteinte bilatérale est généralement incompatible avec la vie en raison d’un oligoamnios ou d’un anamnios entraînant une hypoplasie pulmonaire.44
Épidémiologie
Le rein dysplasique multikystique (MCDK) est l’une des anomalies les plus fréquentes, avec une incidence estimée de 1 pour 1 000 à 4 000 naissances.1,45 La plus forte incidence de rein unique à l’âge adulte est probablement liée à l’involution des MCDK au fil du temps, compte tenu de la faible incidence de la véritable agénésie rénale.5
Physiopathologie
La dysplasie multicystique apparaît avant la formation du néphron, à partir d’une différenciation métanéphrique anormale, d’une anomalie du blastème néphrogène, ou d’une obstruction de haut grade survenant au cours du développement rénal.1 Les kystes sont tapissés d’un épithélium cubique bas entouré de cellules fusiformes et peuvent être remplis d’un liquide protéique ou sanguin.1 Des composants primitifs et dysplasiques, ainsi que du cartilage immature ou des glomérules immatures, peuvent être observés. MCDK est classée en type infundibulo-pyélique avec atrésie du bassinet et de l’uretère, par opposition au type hydronéphrotique, moins fréquent, qui n’implique qu’une atrésie de l’uretère proximal.46 Le rein controlatéral peut présenter une hypertrophie compensatrice.
Évaluation et diagnostic
Actuellement, le MCDK est le plus souvent découvert à l’échographie prénatale, tandis que l’évaluation d’une masse palpable chez un nouveau-né constitue la deuxième présentation la plus fréquente.47
Un diagnostic de MCDK doit être différencié de l’hydronéphrose due à d’autres affections comme l’obstruction de la jonction pyélo-urétérale. Notamment, dans le MCDK, les kystes sont non communicants et les kystes de plus grande taille apparaissent latéralement. En cas d’hydronéphrose, le parenchyme rénal entoure la structure kystique centrale et la forme réniforme typique est conservée.48 Figure 3 et Figure 4 aident à démontrer les difficultés de différencier le MCDK de l’hydronéphrose à l’échographie. Figure 5 illustre des kystes non communicants de tailles variées, typiques du MCDK. Pour préciser davantage, une scintigraphie aux radionucléides peut être réalisée qui montrerait une fixation en cas d’hydronéphrose et, typiquement, une fixation minimale voire absente dans le contexte de MCDK.49
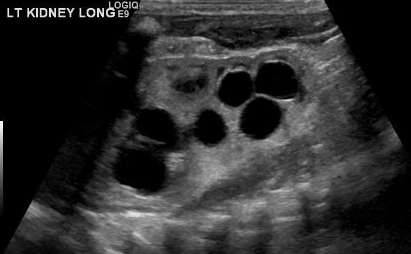
Figure 3 Échographie rénale montrant ce qui pourrait correspondre à plusieurs kystes rénaux dans un MCDK, par opposition à une pyélocalicectasie/hydronéphrose (HDN).
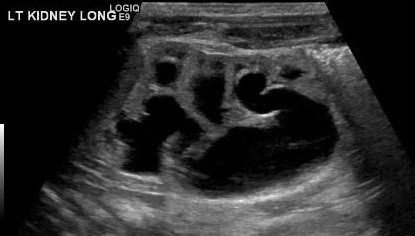
Figure 4 Une autre image d’échographie rénale chez ce même patient, mettant en évidence la continuité entre le bassinet et les calices, compatible avec une pyélocalicectasie/HDN.
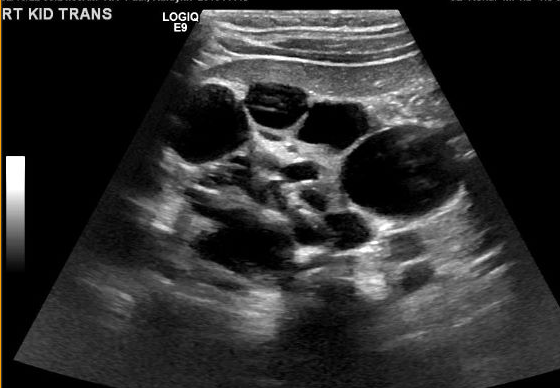
Figure 5 Échographie rénale montrant de multiples kystes non communicants de tailles différentes, compatible avec un MCDK.
Options de traitement, leurs résultats et complications
L’évolution naturelle de la dysplasie rénale multikystique (MCDK) comporte une involution au fil du temps, avec des complications possibles, notamment l’hypertension artérielle et le cancer.5 Cependant, le risque de cette dernière pourrait être faible.50 Historiquement, les reins atteints étaient retirés chirurgicalement par crainte de rares cas de dégénérescence maligne, mais il y a eu une évolution vers une prise en charge non opératoire avec une surveillance échographique des deux reins. Les indications opératoires comprennent des modifications suspectes de développement d’une tumeur de Wilms, un effet de masse, des douleurs, une hypertension artérielle et la préférence des parents.5 Les patients présentant une MCDK et un rein controlatéral normal doivent être rassurés quant à la perspective d’une fonction rénale normale à long terme, à condition que leur rein controlatéral ne soit pas lésé. Ils doivent également être sensibilisés à l’importance des mesures de prévention et d’entretien, comme l’utilisation d’équipements de protection lors de la pratique sportive, ainsi qu’à des choix de vie sains (alimentation et exercice) pour atténuer d’autres causes de maladie rénale (telles que l’hypertension artérielle et le diabète). Pour caractériser le risque de développement d’une maladie rénale chronique, la surveillance peut en outre inclure l’imagerie du rein controlatéral, une urétrocystographie mictionnelle pour dépister un reflux vésico-urétéral (RVU), la surveillance de la pression artérielle et une analyse d’urines à la recherche d’une protéinurie.51
Rein médullaire en éponge
Le rein médullaire en éponge (MSK) est caractérisé par une dilatation des canaux collecteurs distaux, des kystes et des diverticules, tous confinés aux pyramides médullaires.1
Épidémiologie
Le MSK se rencontre principalement au début de l’âge adulte, mais peut également se manifester chez les enfants.52 Comme de nombreuses personnes atteintes de MSK sont asymptomatiques, l’incidence n’est pas clairement définie. Il est plus fréquent chez les personnes ayant des antécédents de lithiase calcique et a été associé à des syndromes congénitaux, tels que le syndrome de Beckwith-Wiedemann, le syndrome d’Ehlers-Danlos, l’anodontie et la maladie de Caroli.1
Physiopathologie
Le MSK est largement considéré comme non héréditaire, cependant, des cas semblant être transmis selon un mode autosomique dominant ont été rapportés, ce qui étaye l’hypothèse selon laquelle le MSK perturbe l’interface entre le bourgeon urétéral et le blastème métanéphrogène.52
Les kystes des canaux collecteurs intrapapillaires dilatés et les kystes médullaires contiennent du matériel calcifié ou desquamatif, confèrent au rein une apparence en éponge, et sont bilatéraux dans 70% des cas.1 Les kystes sont contigus aux tubules collecteurs et sont tapissés par un épithélium de canal collecteur.53
Évaluation et diagnostic
Le diagnostic peut reposer sur des signes urographiques de reins volumineux avec des calcifications occasionnelles, en particulier au niveau des papilles, des tubules papillaires dilatés qui se remplissent de produit de contraste, et un blush papillaire de contraste avec opacification médullaire résiduelle.54 L’évaluation hépatique peut aider à différencier MSK d’ADPKD, si le diagnostic est incertain.1
Options thérapeutiques, leurs résultats et complications
La prise en charge est principalement dictée par le traitement des calculs ou des infections. La prévention des calculs est mise en œuvre, comprenant une augmentation de l’apport hydrique, un régime pauvre en sodium, des diurétiques thiazidiques et du citrate de potassium.52 Les complications associées à la MSK sont liées à l’incidence accrue de la néphrolithiase et à sa morbidité associée.
Néphronophtise
La néphronophtise (NPHP) est une cause fréquente de maladie rénale chronique chez l’enfant, transmise sur un mode autosomique récessif, souvent associée à la néphropathie kystique médullaire (MCKD) car elles présentent des aspects radiologique et histologique similaires ; cependant, la MCKD est transmise sur un mode autosomique dominant, se manifeste plus tardivement (à l’âge adulte) et ne comporte pas d’atteinte extrarénale.5
Épidémiologie
NPHP est classée en forme infantile (1 an), juvénile (13 ans) ou adolescente (19 ans) en fonction de l’anomalie génétique présente. La NPHP infantile conduit à une insuffisance rénale terminale entre 1–3 ans et est associée à des mutations de NPHP2 (également appelées NPHP type 2). La NPHP juvénile, la manifestation la plus fréquente, conduit à une insuffisance rénale terminale à 10–13 ans et est le plus souvent associée à une mutation de NPHP1 (également appelée NPHP type 1). Enfin, la NPHP adolescente est associée à des mutations de NPHP3 (également appelées NPHP type 3).5,55
Pathogénie
Bien qu’elle puisse survenir de façon sporadique, la NPHP est généralement transmise sur un mode autosomique récessif, les mutations délétères homozygotes de NPHP1 sur le chromosome 2 en constituant la cause la plus fréquente.56 De nombreux gènes sont connus pour être associés à la NPHP (voir (Tableau 2)5
Tableau 2 Hétérogénéité génétique de la NPHP.5
| Type | Localisation chromosomique | Gène muté |
|---|---|---|
| Type NPH 1 | 2q13 | NPHP1 |
| Type NPH 2 | 9q22 | NPHP2 |
| Type NPH 3 | 3q22 | NPHP3 |
| Type NPH 4 | 1p36 | NPHP4 |
| Type NPH 5 | 3q21 | NPHP5 |
| Type PH 6 | 12q21 | NPHP6 |
| Type NPH 7 | 16p | NPHP7 |
| Type NPH 8 | 16q | NPHP8 |
| Type NPH 9 | 17q11 | NPHP9 |
| Type NPH 10 | 1q44 | SDCCAG8 |
| Type PH 11 | 8q22 | MKS3 |
| AHI1 | 6q23 | AHI1 |
L’évaluation macroscopique des reins atteints de NPHP peut varier selon le type, mais ils tendent à être de petite taille avec une surface granuleuse. Histologiquement, ils présentent une fibrose interstitielle et une infiltration de cellules mononucléées, ainsi que des remaniements tubulaires variables, incluant une atrophie, un épaississement et une lamellation des membranes basales.57 Les kystes dans la NPHP prennent naissance à partir du tubule contourné distal et des canaux collecteurs.57
Évaluation, diagnostic et complications
Des problèmes de capacité de concentration urinaire peuvent entraîner une polyurie et une polydipsie (déshydratation, nycturie) avec une fuite rénale de sodium qui en résulte; des symptômes d’IRC tels que la fatigue, l’anorexie, un retard de croissance; ainsi que des symptômes d’ostéodystrophie rénale. Une anémie peut être présente de manière disproportionnée par rapport à l’insuffisance rénale. L’HTA est moins fréquente, sauf dans la NPHP de type 2.5,58 L’atteinte extrarénale comprend une fibrose hépatique et portale avec hépatomégalie ainsi que bien d’autres : syndrome de Senior-Løken (NPHP et dégénérescence tapéto-rétinienne), anomalies squelettiques, retard mental, ataxie cérébelleuse, situs inversus, malformation cardiaque, ou d’autres syndromes moins fréquents.5
L’échographie est une méthode courante d’identification, mais elle peut ne pas détecter tous les cas; la tomodensitométrie (TDM) à coupes fines peut détecter de tels cas.58 Des tests génétiques sont disponibles pour le diagnostic, ainsi que l’histologie rénale lorsque l’imagerie est non concluante. Un dépistage hépatique et oculaire est recommandé pour la fibrose hépatique et le syndrome de Senior-Løken.5
Options de traitement et leurs résultats
Il existe peu de traitements efficaces pour la NPHP. La prise en charge actuelle comprend des mesures de soutien et de prévention pour la MRC. La transplantation en cas d’insuffisance rénale est privilégiée, car l’atteinte tubulaire ne survient pas dans le rein du donneur.13
Kystes multiloculaires
Les kystes multiloculaires (néphrome kystique) sont une prolifération néoplasique bénigne survenant chez les adultes et les enfants et qui s’inscrit dans le même spectre que la tumeur de Wilms.
Épidémiologie
La majorité des cas surviennent avant l’âge de 4 ans ou après l’âge de 30 ans, avec une prédominance masculine dans la population pédiatrique.59
Pathogénie
Les lésions sont volumineuses, non communicantes, bien encapsulées par un tissu fibreux, peuvent contenir du tissu embryonnaire, et peuvent comprimer le parenchyme rénal normal.5
Évaluation, diagnostic et options de traitement
Les kystes multiloculaires chez l’enfant se présentent le plus souvent par une masse abdominale palpable.60 L’évaluation est réalisée par échographie ou tomodensitométrie; toutefois, en raison de la difficulté qu’il y a parfois à distinguer les kystes multiloculaires d’une tumeur de Wilms kystique sur la seule base de l’imagerie, le diagnostic définitif peut être posé par un examen histopathologique après néphrectomie ou néphrectomie partielle.60
Maladies kystiques rénales acquises
La maladie kystique rénale acquise (ACKD) désigne les modifications kystiques bilatérales des reins dans le contexte d’une insuffisance rénale terminale et d’azotémie, et non d’une maladie kystique rénale héréditaire.1
Épidémiologie
Des kystes sont retrouvés chez 10 % des patients atteints d’IRT, ce taux augmentant à 44 % et 60 % à 3 et 5 ans après l’initiation de la dialyse.1
Pathogénie
L’étiologie serait médiée par des toxines, l’accumulation de facteurs de croissance, ou une obstruction tubulaire due à la fibrose, à des cristaux d’oxalate, à une occlusion vasculaire ou à une ischémie.1 Les reins sont atteints bilatéralement et généralement de taille inférieure à la normale, tandis que l’histologie montre un épithélium allant de pavimenteux à cubique, similaire à l’épithélium tubulaire distal.4
Évaluation, diagnostic, traitement et complications
La plupart des patients porteurs de kystes sont asymptomatiques, mais peuvent présenter des douleurs et/ou une hématurie dues à des hémorragies intrakystiques spontanées. L’échographie est couramment utilisée pour le diagnostic et la surveillance, tandis que la TDM ou l’IRM peuvent identifier plus aisément les kystes; toutefois, chez les patients en insuffisance rénale terminale, les examens d’imagerie avec produit de contraste IV doivent être gérés avec prudence en raison des effets néphrotoxiques des agents de contraste.1 Les principales complications sont l’hémorragie et la transformation néoplasique.4 Une échographie de surveillance est recommandée, avec néphrectomie si le contexte est évocateur de néoplasie.5 Ainsi, le traitement est géré de manière conservatrice, avec prise en charge des symptômes et adaptation des calendriers de dépistage ou des schémas de dialyse.
Syndromes associés aux maladies kystiques rénales
Les kystes rénaux peuvent être observés dans divers syndromes, dont les plus fréquents peuvent inclure la sclérose tubéreuse à transmission autosomique dominante et la maladie de Von Hippel-Lindau. D’autres, à transmission autosomique récessive, comprennent le syndrome de Meckel, la dystrophie thoracique asphyxiante de Jeune et le syndrome cérébro-hépato-rénal de Zellweger.1
Sclérose tubéreuse
La sclérose tubéreuse est diagnostiquée par la présence soit de deux critères majeurs, soit d’un critère majeur plus deux critères mineurs (voir Tableau 3).61 La présence de kystes rénaux multiples est incluse comme critère mineur.61,62 L’atteinte rénale est une cause importante de morbidité et de mortalité dans la sclérose tubéreuse.63
Tableau 3 Critères majeurs et mineurs de la sclérose tubéreuse de Bourneville.61
| Critères majeurs | Critères mineurs |
|---|---|
| Macules hypomélanotiques (≥3, d’au moins 5 mm de diamètre) | Lésions cutanées « confetti » |
| Angiofibromes (≥3) ou plaque fibreuse céphalique | Fossettes de l’émail dentaire (>3) |
| Fibromes unguéaux (≥2) | Fibromes intra-oraux (≥2) |
| Plaque chagrinée | Plaque achromique rétinienne |
| Hamartomes rétiniens multiples | Kystes rénaux multiples |
| Dysplasies corticales | Hamartomes non rénaux |
| Nodules sous-épendymaires | |
| Astrocytome sous-épendymaire à cellules géantes | |
| Rhabdomyome cardiaque | |
| Lymphangioléiomyomatose (LAM) | |
| Angiomyolipomes (≥2) |
Syndrome de von Hippel-Lindau
Le syndrome de von Hippel-Lindau présente également de nombreuses manifestations, dont le carcinome à cellules rénales et les kystes rénaux sont fréquents, mais rarement observés chez l’enfant.64
Syndrome de Meckel
Cela peut se manifester de diverses manières, notamment par des kystes. La dysplasie kystique est toujours présente.5,65
Conclusions
Les kystes rénaux remplis de liquide se rencontrent dans un vaste groupe hétérogène de maladies kystiques rénales. Bien qu’elles partagent une manifestation commune sous forme de kystes rénaux, ce groupe de maladies se différencie par de nombreux facteurs : mode d’hérédité, âge de début, anomalies rénales ou extrarénales associées, évolution clinique, différences histologiques, aspect en imagerie, entre autres. L’identification précise d’un processus pathologique peut être difficile en raison d’une symptomatologie chevauchante. Les techniques d’imagerie, en particulier l’échographie, constituent depuis longtemps un moyen principal d’identifier un grand nombre de ces maladies. Les options de tests génétiques deviennent de plus en plus courantes et accessibles.
La poursuite de la recherche conduit à des avancées en sciences fondamentales et en clinique, permettant d’identifier de nouvelles maladies, la physiopathologie, des modalités diagnostiques et des options thérapeutiques.
Points clés
- La distinction entre un kyste simple et un diverticule caliciel peut être difficile, mais elle est particulièrement importante car un diverticule caliciel est beaucoup plus susceptible de provoquer des symptômes et de nécessiter une intervention. Le diagnostic peut être facilité au moyen d’une imagerie en coupes avec contraste IV et d’images en phase tardive.
- Il est également important de distinguer le MCDK d’une hydronéphrose sévère, comme dans le contexte d’une obstruction de la jonction pyélo-urétérale (UPJ). La prise en charge du MCDK est en grande partie observationnelle, tandis qu’il est plus probable qu’une intervention soit nécessaire en cas d’hydronéphrose ou d’obstruction de l’UPJ. Le diagnostic peut être facilité grâce à l’imagerie aux radionucléides.
- La prise en charge de certaines maladies kystiques rénales, telles que l’ADPKD et l’ARPKD, doit être réalisée en partenariat avec la néphrologie afin de favoriser des soins médico-rénaux optimaux.
- La caractérisation des kystes par l’imagerie prénatale aide souvent à différencier les maladies possibles (voir Figure 6 ci-dessous).13

Figure 6 Différenciation des kystes par l’imagerie prénatale.13
Lectures recommandées
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
Références
- Pope JC. Renal Dysgenesis and Cystic Disease of the Kidney. 12th ed., Elsevier Saunders; 2021, DOI: 10.1055/a-1307-2419.
- Glassberg KI, Stephens FD, Lebowitz RL. Renal dysgenesis and cystic disease of the kidney: a report of the Committee on Terminology, Nomenclature and Classification, Section on Urology, American Academy of Pediatrics. J Urol Oct 1987; 138 (4 Pt 2): 1085–1092. DOI: 10.1016/s0022-5347(17)43510-5.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364 (16): 1533–1543. DOI: 10.1056/NEJMra1010172.
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol Jan 2006; 13 (1): 26–56. DOI: 10.1097/01.pap.0000201831.77472.d3.
- George RP, Greenbaum LA. Cystic Kidney Disease. 6th ed., CRC Press; 2017, DOI: 10.1201/9781315113982.
- Cole BR, Conley SB, Stapleton FB. Polycystic kidney disease in the first year of life. J Pediatr Nov 1987; 111 (5): 693–699. DOI: 10.1016/s0022-3476(87)80244-5.
- Gabow PA. Polycystic kidney disease: clues to pathogenesis. Kidney Int Dec 1991; 40 (6): 989–996. DOI: 10.1038/ki.1991.306.
- Proesmans W, Damme B, Casaer P, Marchal G. Autosomal dominant polycystic kidney disease in the neonatal period: association with a cerebral arteriovenous malformation. Pediatrics Dec 1982; 70 (6): 971–975. DOI: 10.1542/peds.70.6.971.
- Ong AC, Wheatley DN. Polycystic kidney disease–the ciliary connection. Lancet Mar 2003; 361 (9359): 774–776. DOI: 10.1016/S0140-6736(03)12662-1.
- Rossetti S, Consugar MB, Chapman AB. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol Jul 2007; 18 (7): 2143–2160. DOI: 10.1681/ASN.2006121387.
- Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol Oct 2004; 15 (10): 2528–2536. DOI: 10.1097/01.ASN.0000141055.57643.E0.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet Apr 1994; 343 (8901): 824–827. DOI: 10.1016/s0140-6736(94)92026-5.
- Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am J Kidney Dis 2021; 78 (1): 125–141. DOI: 10.1053/j.ajkd.2020.10.021.
- Grantham JJ. Polycystic kidney disease: hereditary and acquired. Adv Intern Med 1993; 38: 409–420.
- Schrier R, McFann K, Johnson A. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol Jul 2002; 13 (7): 1733–1739. DOI: 10.1097/01.asn.0000018407.60002.b9.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med Oct 1990; 323 (16): 1091–1096. DOI: 10.1056/NEJM199010183231602.
- Alzarka B, Morizono H, Bollman JW, Kim D, Guay-Woodford LM. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Front Pediatr 2017; 5 (80). DOI: 10.3389/fped.2017.00080.
- Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers 2018; 4 (1). DOI: 10.1038/s41572-018-0047-y.
- Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatr Nephrol Jan 1989; 3 (1): 43–49. DOI: 10.1007/BF00859625.
- Ward CJ, Hogan MC, Rossetti S. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet Mar 2002; 30 (3): 259–269. DOI: 10.1038/ng833.
- Onuchic LF, Furu L, Nagasawa Y. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet May 2002; 70 (5): 1305–1317. DOI: 10.1086/340448.
- Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol Jun 1997; 11 (3): 302–306. DOI: 10.1007/s004670050281.
- Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics Sep 2014; 134 (3). DOI: 10.1542/peds.2013-3646.
- Blazer S, Zimmer EZ, Blumenfeld Z, Zelikovic I, Bronshtein M. Natural history of fetal simple renal cysts detected in early pregnancy. J Urol 1999; 162 (3 Pt 1): 812–814. DOI: 10.1097/00005392-199909010-00066.
- McHugh K, Stringer DA, Hebert D, Babiak CA. Simple renal cysts in children: diagnosis and follow-up with US. Radiology Feb 1991; 178 (2): 383–385. DOI: 10.1148/radiology.178.2.1987597.
- Bosniak MA. The current radiological approach to renal cysts. Radiology Jan 1986; 158 (1): 1–10. DOI: 10.1148/radiology.158.1.3510019.
- Lee J, Darcy M. Renal cysts and urinomas. Semin Intervent Radiol. Dec 2011; 28 (4): 380–391. DOI: 10.1055/s-0031-1296080.
- Steinhardt GF, Slovis TL, Perlmutter AD. Simple renal cysts in infants. Radiology May 1985; 155 (2): 349–350. DOI: 10.1148/radiology.155.2.3885305.
- Gabow PA, Kimberling WJ, Strain JD, Manco-Johnson ML, Johnson AM. Utility of ultrasonography in the diagnosis of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol Jan 1997; 8 (1): 105–110. DOI: 10.1681/ASN.V81105.
- Peard L, Gargollo P, Grant C. Validation of the modified Bosniak classification system to risk stratify pediatric cystic renal masses: An international, multi-site study from the pediatric urologic oncology working group of the societies for pediatric urology. J Pediatr Urol 2022; 18 (2). DOI: 10.1016/j.jpurol.2021.12.001.
- Bayram MT, Alaygut D, Soylu A, Serdaroğlu E, Cakmakçı H, Kavukçu S. Clinical and radiological course of simple renal cysts in children. Urology Feb 2014; 83 (2): 433–437. DOI: 10.1016/j.urology.2013.08.055.
- Karmazyn B, Tawadros A, Delaney LR. Ultrasound classification of solitary renal cysts in children. J Pediatr Urol Jun 2015; 11 (3). DOI: 10.1016/j.jpurol.2015.03.001.
- O’Kelly F, McAlpine K, Abdeen N, Keays MA, Leonard MP, Guerra LA. The Prevalence, Clinicodemographics, and Outcomes of Incidental and Symptomatic Renal Cysts in a Pediatric Cohort Undergoing Ultrasonography. J Urol 2019; 202 (2): 394–399. DOI: 10.1097/JU.0000000000000264.
- Rediger C, Guerra LA, Keays MA. Renal cyst evolution in childhood: a contemporary observational study. J Pediatr Urol Apr 2019; 15 (2). DOI: 10.1016/j.jpurol.2019.01.006.
- Wulfsohn MA. Pyelocaliceal diverticula. J Urol Jan 1980; 123 (1): 1–8. DOI: 10.1016/s0022-5347(17)55748-1.
- Estrada CR, Datta S, Schneck FX, Bauer SB, Peters CA, Retik AB. Caliceal diverticula in children: natural history and management. J Urol Mar 2009; 181 (3): 1311. DOI: 10.1016/j.juro.2008.10.043.
- Waingankar N, Hayek S, Smith AD, Okeke Z. Calyceal diverticula: a comprehensive review. Rev Urol 2014; 16 (1): 29–43.
- Sahin H, Sarioglu FC, Alaygut D, Akdogan AI, Pekcevik Y. Differentiation of simple renal parenchymal cyst and calyceal diverticulum. Pediatr Int May 2020; 62 (5): 615–623. DOI: 10.1111/ped.14127.
- Casale P, Grady RW, Feng WC, Joyner BD, Mitchell ME. The pediatric caliceal diverticulum: diagnosis and laparoscopic management. J Endourol Sep 2004; 18 (7): 668–671. DOI: 10.1089/end.2004.18.668.
- Long CJ, Weiss DA, Kolon TF, Srinivasan AK, Shukla AR. Pediatric calyceal diverticulum treatment: An experience with endoscopic and laparoscopic approaches. J Pediatr Urol Aug 2015; 11 (4). DOI: 10.1016/j.jpurol.2015.04.013.
- Sripathi V, Mitra A, Padankatti RL, Ganesan T. Robotic treatment of a type 2 calyceal diverticulum in a child: is suture closure and marsupialisation enough for a good outcome? J Robot Surg Dec 2018; 12 (4): 727–730. DOI: 10.1007/s11701-017-0758-1.
- Ding X, Xu ST, Huang YH. Management of symptomatic caliceal diverticular calculi: Minimally invasive percutaneous nephrolithotomy versus flexible ureterorenoscopy. Chronic Dis Transl Med Dec 2016; 2 (4): 250–256. DOI: 10.1016/j.cdtm.2016.11.016.
- Monga M, Smith R, Ferral H, Thomas R. Percutaneous ablation of caliceal diverticulum: long-term followup. J Urol Jan 2000; 163 (1): 28–32. DOI: 10.1016/s0022-5347(05)67965-7.
- Gilbert-Barness E, Potter EL. Respiratory system. 2nd ed., DOI: 10.1001/jama.1997.03540320078045.
- D’Alton M, Romero R, Grannum P, DePalma L, Jeanty P, Hobbins JC. Antenatal diagnosis of renal anomalies with ultrasound. IV Bilateral Multicystic Kidney Disease Am J Obstet Gynecol Mar 1986; 154 (3): 532–537. DOI: 10.1016/0002-9378(86)90597-1.
- Kalyoussef E, Hwang J, Prasad V, Barone J. Segmental multicystic dysplastic kidney in children. Urology Nov 2006; 68 (5). DOI: 10.1016/j.urology.2006.06.024.
- Felson B, Cussen LJ. The hydronephrotic type of unilateral congenital multicystic disease of the kidney. Semin Roentgenol Apr 1975; 10 (2): 113–123. DOI: 10.1016/0037-198x(75)90035-8.
- Welch TR, Wacksman J. The changing approach to multicystic dysplastic kidney in children. J Pediatr Jun 2005; 146 (6): 723–725. DOI: 10.1016/j.jpeds.2005.02.027.
- Sanders RC, Hartman DS. The sonographic distinction between neonatal multicystic kidney and hydronephrosis. Radiology Jun 1984; 151 (3): 621–625. DOI: 10.1148/radiology.151.3.6718720.
- Roach PJ, Paltiel HJ, Perez-Atayde A, Tello RJ, Davis RT, Treves ST. Renal dysplasia in infants: appearance on 99mTc DMSA scintigraphy. Pediatr Radiol 1995; 25 (6): 472–475. DOI: 10.1007/BF02019071.
- Narchi H. Risk of Wilms’ tumour with multicystic kidney disease: a systematic review. Arch Dis Child Feb 2005; 90 (2): 147–149. DOI: 10.1136/adc.2004.051243.
- Mansoor O, Chandar J, Rodriguez MM. Long-term risk of chronic kidney disease in unilateral multicystic dysplastic kidney. Pediatr Nephrol Apr 2011; 26 (4): 597–603. DOI: 10.1007/s00467-010-1746-0.
- Gambaro G, Danza FM, Fabris A. Medullary sponge kidney. Curr Opin Nephrol Hypertens. Jul 2013; 22 (4): 421–426. DOI: 10.1097/MNH.0b013e3283622b86.
- Bernstein J. The classification of renal cysts. Nephron 1973; 11 (2): 91–100. DOI: 10.1159/000180222.
- Gedroyc WM, Saxton HM. More medullary sponge variants. Clin Radiol Jul 1988; 39 (4): 423–425. DOI: 10.1016/s0009-9260(88)80292-7.
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol Jan 2009; 20 (1): 23–35. DOI: 10.1681/ASN.2008050456.
- Hildebrandt F, Otto E, Rensing C. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet Oct 1997; 17 (2): 149–153. DOI: 10.1038/ng1097-149.
- Sherman FE, Studnicki FM, Fetterman G. Renal lesions of familial juvenile nephronophthisis examined by microdissection. Am J Clin Pathol Apr 1971; 55 (4): 391–400. DOI: 10.1093/ajcp/55.4.391.
- Elzouki AY, al-Suhaibani H, Mirza K, al-Sowailem AM. Thin-section computed tomography scans detect medullary cysts in patients believed to have juvenile nephronophthisis. Am J Kidney Dis Feb 1996; 27 (2): 216–219. DOI: 10.1016/s0272-6386(96)90543-0.
- Eble JN, Bonsib SM. Extensively cystic renal neoplasms: cystic nephroma, cystic partially differentiated nephroblastoma, multilocular cystic renal cell carcinoma, and cystic hamartoma of renal pelvis. Semin Diagn Pathol Feb 1998; 15 (1): 2–20.
- Castillo OA, Boyle ET, Kramer SA. Multilocular cysts of kidney. A study of 29 patients and review of literature. Urology Feb 1991; 37 (2): 156–162. DOI: 10.1016/0090-4295(91)80214-r.
- Northrup H, Krueger DA, ITSCC G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol Oct 2013; 49 (4): 243–254. DOI: 10.1016/j.pediatrneurol.2013.08.001.
- Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol Dec 1998; 13 (12): 624–628. DOI: 10.1177/088307389801301206.
- Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc Aug 1991; 66 (8): 792–796. DOI: 10.1016/s0025-6196(12)61196-3.
- Maddock IR, Moran A, Maher ER. A genetic register for von Hippel-Lindau disease. J Med Genet Feb 1996; 33 (2): 120–127. DOI: 10.1136/jmg.33.2.120.
- Salonen R. The Meckel syndrome: clinicopathological findings in 67 patients. Am J Med Genet Aug 1984; 18 (4): 671–689. DOI: 10.1002/ajmg.1320180414.
Dernière mise à jour: 2025-09-22 07:59